Volume 32, Number 4—April 2026
Research
Geographically Distinct Circulation of Genotype II and III St. Louis Encephalitis Virus, Texas, USA, 2009–2024
Cite This Article
Citation for Media
Abstract
We conducted a retrospective genomic surveillance study of St. Louis encephalitis virus (SLEV) in Texas, USA, to determine the genotypes circulating in the region. By using a custom tiled-amplicon assay with Oxford Nanopore sequencing, we generated 63 genomes from SLEV-positive mosquito pools and viral isolates collected during 2009–2024. Phylogenomic analysis revealed temporal overlap of genotype II and III circulation, but with distinct geographic segregation. Genotype II was confined to Gulf Coast counties with sustained local transmission, whereas genotype III was only in north and west Texas, but with persistent circulation and repeated introductions. We identified the earliest known US genotype III sequences, although their phylogenetic placement leaves the entry point of genotype III into the United States unresolved. Our findings emphasize the need for clinical vigilance in West Texas, where SLEV and West Nile virus co-circulate, and suggest the Gulf Coast may be buffered against foreign genotype introduction.
St. Louis encephalitis virus (SLEV) is a recently reemerged mosquitoborne orthoflavivirus found in the Americas (1). SLEV is maintained in an enzootic cycle between mosquitoes (primarily Culex spp.) and avian reservoirs; mammals are dead-end hosts (2–5). Since the virus’s first identification in 1933 in St. Louis, Missouri, USA (6), focal SLEV outbreaks have been reported throughout the United States; disease manifestations range from nonspecific influenza-like symptoms to severe, potentially fatal, neurologic complications (7–9). After West Nile virus (WNV) was introduced to the United States in 1999 (10), a marked decrease in SLEV-positive mosquito pools and human cases occurred in areas where WNV was present (11,12). Both viruses, members of the Japanese encephalitis virus serocomplex, confer cross-reactive immunity in avian hosts (13–15), and the decrease in SLEV occurrence potentially is driven by ecologic competition between WNV and SLEV in reservoir populations because of cross-protective immune responses (11,14). SLEV reemerged in the United States after the introduction of a new genotype to the southwestern United States.
Eight genotypes of SLEV exist. Genotypes I, II, IV, and V are historically found in North America, and genotypes III, V, VI, VII, and VIII are found in South America (16,17). SLEV human cases and outbreaks have occurred in North America, and none had been reported in South America (1,7) until 2005, when an outbreak caused human fatalities in Argentina (18). Subsequent 2010 and 2011 outbreaks in Argentina were identified as genotype III, which previously had not been associated with human cases (1). In 2015, the first concurrent outbreak of WNV and SLEV occurred in Arizona, USA, prompting SLEV surveillance efforts (19,20). That same year, genotype III SLEV was detected in California, USA, after nearly a decade without reported activity despite ongoing monitoring. Retrospective analyses identified the viruses in both states as genotype III and revealed a 2014 mosquito pool from Arizona that was positive for SLEV and WNV (20). Genotype III outbreaks in California and Arizona had a case-fatality rate of ≈8% (8,19–22). Further genomic surveillance indicated that SLEV genotype III replaced all other SLEV genotypes in the western United States (21,22) but analyzed samples from as far east as El Paso, Texas, USA.
Figure 1

Figure 1. Annual SLEV and WNV mosquito and human case activity from study of circulation of genotype II and III SLEV, Texas, USA, 2002–2024. A) Annual number of reported SLEV-positive and WNV-positive...
SLEV was a prevalent mosquitoborne virus in Texas, particularly along the Gulf Coast, before the introduction of WNV in 2002; SLEV cases and mosquito activity declined in the region after WNV was established (1,23–26). During the past 20 years, sporadic reports of SLEV human cases and SLEV-positive mosquito pools occurred throughout Texas (Figure 1, panels A–D). However, only 2 full-length Texas SLEV genomes (GenBank accession nos. EF158052.1 and MN233324.1) have been detected, and no information is available regarding which genotypes are currently circulating. To address those deficiencies and determine whether genotype III is widespread throughout Texas, we performed a retrospective genomic surveillance study analyzing SLEV genomes from SLEV-positive mosquito pools during 2009–2024.
We have described in detail the methods used in this study (Appendix). In addition, a GitHub repository for this work (https://github.com/kneubehl/Texas-SLEV-Genome-Surveillance) (27) contains other ancillary items (e.g., scripts).
Mosquito Pools
Mosquito pools were provided by the Texas Department of Safety and Health Services, Harris County Public Health Mosquito and Vector Control Division, or Texas Tech University’s Biologic Threat Research Laboratory, each having their own respective collection and testing strategies (Appendix). We summarized mosquito pool metadata (Appendix Table 1) (27). Because virus screening modalities varied on the basis of a mosquito pool’s origin, we screened all pools by using a real-time reverse transcription PCR (rRT-PCR) to confirm positivity.
Virus Isolation
We performed virus isolation by incubating mosquito pool homogenate with Vero CCL81 cells in T75 flasks. When cytopathic effects were apparent or after 7 days, whichever came first, we clarified cell culture supernatants by using centrifugation. We stored an aliquot of supernatant in 2x DNA/RNA Shield (Zymo, https://www.zymoresearch.com) for RNA extraction. We obtained 4 SLEV isolates through BEI Resources at the National Institutes of Health National Institute of Allergy and Infectious Diseases (V07457, V08449, V08458, and TXAR 9-6038). We summarized virus isolate metadata (Appendix Table 2) (27).
RNA Isolation and SLEV rRT-PCR Testing
We isolated total RNA and DNA from samples by using a modified Zymo Quick-DNA/RNA Pathogen Miniprep protocol (Appendix). We tested all samples for the presence of SLEV RNA by using a modified rRT-PCR protocol (28) (Appendix).
Tiled-Amplicon Sequencing Primer Development
We designed a tiled-amplicon primer scheme to amplify the entire coding sequence of the SLEV genome (97.4% of the entire genome sequence) by using the Olivar tool with SLEV genomes sourced from GenBank. We used DQ525916.1 as the reference genome (27,29) (Appendix Table 3).
Virus Genome Amplification, Sequencing, and Assembly
We generated SLEV tiled amplicons by using reverse transcription -PCR (Appendix). We prepared sequencing libraries by using the SQK-RBK114.96 kit (Oxford Nanopore Technologies, https://www.nanoporetech.com) and sequenced them with an R10.4.1 flow cell (Oxford Nanopore), per manufacturer’s instructions, on a MinION Mk1B (Oxford Nanopore) or PromethION P2 solo (Oxford Nanopore). We generated consensus genome assemblies by sample with the ViralRecon nextflow pipeline with the ARTIC minion pipeline (https://zenodo.org/records/7764938) option with modifications (Appendix). Sequencing data and genomes generated in this study are available through BioProject PRJNA1273178.
Maximum-Likelihood Analyses of All SLEV Genotypes and SLEV Genotype II Envelope
To determine the phylogenetic placement of our SLEV assemblies, we inferred a maximum-likelihood tree of all SLEV genotypes with available genomes on GenBank. We obtained SLEV genomes from GenBank, and we analyzed only genomes with >70% completeness. We aligned the nucleotide sequences by using MAFFT (30). We inferred a maximum-likelihood tree by using IQ-TREE2 (31,32). We used the same process for SLEV genotype II envelope sequences. We visualized and annotated the phylogenies in R (The R Project for Statistical Computing, https://www.r-project.org) and Inkscape (Inkscape, https://inkscape.org). Newick tree files for each phylogeny are available (27).
Bayesian Phylogenetic Analysis of SLEV Genotype III
We assessed the maximum-likelihood tree of all SLEV genotype III genomes with root-to-tip genetic divergence and time of sampling regression by using TempEst (33). That analysis determined a temporal signal that warranted generating a timetree (correlation coefficient 0.8284). We used BEAST for Bayesian phylogenetic analysis (34). We used an uncorrelated relaxed clock model, a general time reversible plus empirical base frequencies plus gamma-distributed rate heterogeneity with 4 categories substitution model, and a Bayesian skyline with a 2-groups tree prior and a Markov chain Monte Carlo chain length of 100 million with sampling every 10,000 trees to infer the timetree. We ran 2 independent MCMC chains, and we confirmed convergence with Tracer before combining with LogCombiner (34), using a 10% burn-in for each log and tree file. We generated the maximum clade credibility tree with TreeAnnotator (34). The XML generated by BEAUti for running BEAST and the nexus file generated by TreeAnnotator have been described previously (27). We visualized and annotated the phylogenies by using R and Inkscape.
Genotyping SLEV-Positive Cx. quinquefasciatus and Cx. tarsalis Mosquito Pools
Figure 2
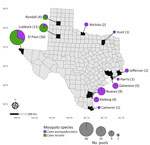
Figure 2. St. Louis encephalitis virus–positive mosquito pool samples from study of circulation of genotype II and III St. Louis encephalitis virus, by county of origin, Texas, USA, 2002–2024. Each county of...
Our sample set included 75 SLEV-positive mosquito pools from 11 counties in Texas and 4 previously generated Texas SLEV isolates (Figure 2). Each pool consisted of Cx. tarsalis (36 pools) or Cx. quinquefasciatus (39 pools) collected during 2009–2024. Cx. tarsalis pools came from northwestern Texas, whereas Cx. quinquefasciatus pools were collected throughout the state (Figure 2). Our sample set included 5 mosquito pools positive for SLEV and WNV from El Paso County (4 pools) and Lubbock County (1 pool).
We used a tiled-amplicon sequencing approach for genome sequencing of SLEV. We also attempted virus isolation on all 64 mosquito pools not in a preservative to amplify the virus in case sequencing attempts from the mosquito pool RNA failed. Of the 123 samples tested (75 mosquito pools, 44 successful virus isolations, and 4 preexisting virus isolates), 63 unique SLEV genomes had >70% genome completeness. We assembled 37 genomes directly from mosquito pools, 22 from virus isolates that yielded more complete genomes than their corresponding mosquito pools, and 4 from previously available isolates. Six other mosquito pools had genome assemblies with <70% completeness; we genotyped those but did not use them for phylogenetic analysis. We plotted virus genome completeness with each sample’s cycle threshold (Ct) value determined by rRT-PCR for all 123 sequencing attempts (Appendix Figure 1) (27). The median Ct for samples that were successful (>70% genome completeness) was 22.7 (range 16.2–29.5). We summarized read depth across the genome from representative samples spanning different Ct values and genome recovery (Appendix Figure 2). We also summarized genome completeness for each sample (Appendix Tables 1, 2) (27).
Phylogenetic Analysis of SLEV Genomes
Figure 3

Figure 3. Maximum-likelihood phylogenic analysis of all St. Louis encephalitis virus genomes from study of circulation of genotype II and III St. Louis encephalitis virus, Texas, USA, 2009–2024. Maximum-likelihood inferred tree shows...
Figure 4

Figure 4. SLEV genotype distribution from study of circulation of genotype II and III SLEV, by year and county, Texas, USA, 2009–2024. A) Dot plot of number of sequenced SLEV-positive mosquito pools...
Maximum-likelihood phylogenetic analyses defined the genotypes of our samples and their genetic relatedness to 276 previously published SLEV genomes (63 of our assemblies and 213 GenBank assemblies). We genotyped the 6 samples with a genome completeness <70% but >5% by using BLASTn (https://blast.ncbi.nlm.nih.gov) (Appendix Table 4) (27). Our analyses indicated that genotypes II (n = 26) and III (n = 43) were present in Texas concurrently (Figure 3). Genotype II was detected in 2009, 2010, 2013, 2014, 2016, 2020, 2021, and 2023; genotype III was present in 2014, 2015, and 2017–2024 (Figure 4, panel A). Although temporal overlap of genotypes occurred, we noted spatial segregation: genotype II in coastal counties (Cameron, Harris, Galveston, Jefferson, Kleberg, and Nueces) and genotype III in northern and western counties (El Paso, Hunt, Lubbock, Randall, and Wichita) (Figure 4, panel B).
Phylogenetic Analysis of Genotype II Envelope Gene
Figure 5

Figure 5. Maximum-likelihood phylogenic analysis of St. Louis encephalitis virus genotype IIA and IIB envelope gene sequences from study of circulation of genotype II and III St. Louis encephalitis virus, Texas, USA....
Because only a limited number of whole-genome sequences of genotype II SLEV were publicly available (n = 28), we used the envelope gene for phylogenetic analysis to increase the sample size spanning a broader timeframe (n = 61 publicly available sequences) (Figure 5). That analysis indicated that these samples fell within 2 clusters in genotype IIB. Our samples all share a basal node from a 1991 Harris County sample, but another more derived sample from 1983 indicates persistence in the region since at least the 1980s. Harris and Nueces County samples from 2013 (n = 5) clustered together in a position basal to the other samples from our dataset. Other samples from Galveston, Harris, Jefferson, Kleberg, and Nueces Counties (n = 20) span from 2009 to 2023 and clustered into a polytomy with other previously sequenced samples from Harris and Jefferson Counties, including a sample from Louisiana. This clade has a basal node of 2 samples from Harris County collected in 2003. Samples from Kleberg, Nueces, and Galveston Counties (n = 14) from 2020 and 2021 all clustered together, to the exclusion of the 6 remaining samples from Harris, Jefferson, and Nueces Counties that had collection dates spanning from 2009 to 2023. We compared the pairwise percentage identity of each genotype II sequence and determined that, across both genotype IIA and IIB, they differed in sequence identity by at most 4.5% (Appendix Figure 2) (27). Within genotypes IIA and IIB, we found a 2.7% difference in sequence identity in the envelope gene.
Bayesian Phylogenetic Analysis of Genotype III Genomes
Figure 6

Figure 6. Bayesian phylogenetic analysis of SLEV genotype III from Texas and other states from study of circulation of genotype II and III St. Louis encephalitis virus, Texas, USA. Maximum clade credibility...
Given the epidemic potential of genotype III and to better understand introduction events and divergence times, we performed a more rigorous phylogenetic analysis by using Bayesian methods. TempEST (33) identified sufficient temporal signal (correlation coefficient 0.8284) in our genotype III dataset by using the root-to-tip distance compared with the date of collection from a maximum-likelihood tree inferred from the 216 genotype III genomes available (38 of our assemblies and 178 GenBank assemblies). The Bayesian time tree recapitulated a similar topology to a previous analysis (21), which showed US samples forming 2 clades (clades 1 and 2) (Figure 6); the node of the time to most recent common ancestor (tMRCA) of genotype III in the United States was March 2013 (95% highest posterior density [HPD] interval December 2011–October 2014).
Clade 1 primarily contained US samples collected during 2014–2024 and included the country’s earliest known genotype III sample, a SLEV- and WNV-positive Cx. tarsalis pool collected in July 2014 from Lubbock County. Clade 1 also contained 3 additional SLEV- and WNV-positive mosquito pools from El Paso County. Furthermore, this clade contained Lubbock County samples from 2024 with a well-supported basal node from a 2017 El Paso sample, suggesting sustained circulation of genotype III virus in West Texas. Other El Paso County samples from 2019 and 2021 did not form well-supported subclades, indicating possible introduction events from other regions. The newly sequenced 2014 Texas samples were not basal to the other US genotype III sequences, leaving the origin of genotype III introduction into the US unresolved.
Clade 2 consisted of samples from 2017 onward. The Bayesian time tree recapitulated subclades 2A, which were largely California samples (tMRCA June 2016 [95% HPD interval November 2015–March 2017]), and 2B (tMRCA December 2014 [95% HPD interval July 2014–March 2015]), which generally were Arizona samples. The Texas samples were within subclade 2B, clustering with the Arizona samples and subsequently forming 2 moderately supported subclades we called 2B1 (posterior probability 0.7898) and 2B2 (posterior probability 0.7933). Subclade 2B1 (tMRCA June 2016 [95% HPD interval August 2015–March 2017]) had limited sampling over multiple years from El Paso, Wichita, and Lubbock Counties, so determining persistence from year to year is limited. The Texas samples in this subclade were related to samples collected from Arizona, indicating an introduction from that region. The Texas samples bifurcated into sister clades; El Paso County samples clustered to the exclusion of Wichita and Lubbock Counties, although both clades shared a basal node from a 2021 El Paso County sample. In both groups, samples from >2 years are present (2021 and 2022 from El Paso County and 2023 and 2024 from Lubbock County), suggesting persistence of those viruses. Subclade 2B2 (tMRCA February 2015 [95% HPD interval November 2014–May 2015]) is a radiation with poorly supported virus population structure. This subclade is dominated by samples from Arizona; only 5 Texas samples (El Paso County 2019 and 2020) clustered into this group. These samples probably indicate introduction events into El Paso County from outside Texas that failed to persist or have remained undetected in subsequent years. El Paso County (2019 and 2021) and Lubbock County (2024) had clade 1 and clade 2 viruses, indicating SLEV genetic diversity within the same county. Collectively, the Bayesian phylogenetic analysis indicated repeated introduction events of genotype III into Texas and persistence of phylogenetically distinct viruses falling into clades 1 and 2.
This study highlights the importance of banking and sharing mosquito pool samples and collaborative surveillance efforts. Our retrospective study identified circulation of SLEV genotypes II and III in Texas during 2009–2024 and showed geographic segregation of each genotype, addressing a major gap in knowledge of genotype distribution in the state. Texas has 254 counties each with different mosquito sampling and pathogen testing algorithms, including some with no sampling or screening. Our sample size and scope were limited to those areas that tested and reported SLEV-positive mosquitoes. Of the 173 SLEV-positive mosquito pools reported since 2002 by Texas Department of Safety and Health Services, 43.3% (n = 75) were available, and 74 of those pools were collected after 2012. Because of availability of banked samples, our analysis did not include pools from the northern and western Texas counties before the introduction of genotype III, and it remains unknown which genotype or genotypes circulated immediately before the introduction. Further surveillance is needed to enhance the temporal resolution and improve classifications of introduction events from established virus lineages for SLEV and other arboviruses.
Genotypes I, II, and V SLEV historically are reported in Texas, and genotype II has dominated the Gulf Coast region since the 1980s (16,35). In line with previous findings, our results indicate that genotype IIB is still circulating >40 years after becoming dominant in the region. We detected different genotype IIB variants circulating in Nueces and Harris Counties at different times. The 2013 samples from Nueces and Harris Counties shared a recent common ancestor with samples from the same counties from 1991, 1983, and 2002, suggesting that the 2013 samples stem from an older lineage of genotype II that had not been sampled for at least a decade. No genotype III–positive samples were detected in the Gulf Coast region (Cameron, Galveston, Harris, Jefferson, Kleberg, and Nueces Counties) during 2009–2024 but were present during 2014–2024 in northern and western Texas counties (El Paso, Hunt, Lubbock, and Randall). These genotype III samples included the earliest known examples of genotype III virus in the United States. Our new data indicate that the genotype III introduction timeframe was December 2011–October 2014, differing slightly from previous reports of September 2011–May 2014 (21) and August 2012–October 2013 (22), suggesting that genotype III was circulating in the United States well before it was first detected in 2014. Our 2014 Texas samples were not basal to the other genotype III samples, indicating that the introduction site to the United States is still unknown. The earliest genotype III sample from Lubbock County was related to samples collected in Oregon and Idaho in 2017, indicating that Texas may have been an early area of genotype III introduction and that genotype III may have been exported to parts of the West Coast. We detected phylogenetically related viruses across years in clade 1 (early introduction) and clade 2 (later introduction), indicating establishment in Texas and greater viral diversity compared with Arizona and California, where clade 1 circulation is no longer reported (21,22). We detected introduction events with no direct ancestors from Texas in all clades, suggesting that West Texas is a site of continuous introduction events of SLEV genotype III.
The importation and circulation of new SLEV genotypes in West Texas, particularly in El Paso County, is not a new phenomenon. In the early 2000s, genotype VA circulated in El Paso County, whereas genotype IIB circulated in the Gulf Coast region (35). The El Paso isolates were genetically similar to genotype VA from South America that was contemporaneously circulating in California (16,35). Others hypothesized that cooler regions, such as El Paso County, require annual reintroduction of SLEV because overwintering does not appear to be effective for virus maintenance; however, in the subtropical Texas Gulf Coast region, SLEV can overwinter by unknown means (35). In contrast, our data supports year-to-year persistence of related genotype III viruses in El Paso County and potential SLEV overwintering in the county. Given the dispersal of SLEV occurs South-to-North from Central and South America to North America (36), and given the importance of the Texas Gulf Coast region in the Central Flyway for migratory birds from Central and South America (37), we would expect the Gulf Coast region rather than West Texas to be a major site for SLEV introductions. Further work is necessary to determine which migratory birds in West Texas are facilitating SLEV introduction events and how genotype III is overwintering in West Texas.
The observation that western and northern Texas counties harbor a different circulating genotype than Gulf Coast counties was previously reported and probably is likely driven by differences in rural–urban transmission cycles shaped by Culex mosquito distribution (16,35). Cx. tarsalis mosquitoes, typically involved in the rural transmission cycle of SLEV and associated with sporadic human infections (38,39), are better adapted to habitats in western Texas (40). Cx. quinquefasciatus mosquitoes, the primary vector in the urban transmission cycle and a driver of SLEV epidemics (25,26,41,42), are better adapted to habitats along the Gulf Coast region (40). Our samples generally align with this geographic dichotomy: all West Texas pools contained Cx. tarsalis mosquitoes, and 56% of Gulf Coast pools contained Cx. quinquefasciatus mosquitoes. SLEV genotypes and variants circulating in West Texas’ rural cycle can invade and establish urban cycles in more populated parts of Texas, resulting in SLEV outbreaks. Spillover from the SLEV rural cycle in West Texas to the Dallas metropolitan area in the 1960s probably resulted in outbreaks (41). Although we detected a SLEV genotype III–positive Cx. quinquefasciatus mosquito pool from 2014 in Hunt County, bordering the Dallas metropolitan area, this virus probably was an introduction from outside of Texas. Continued surveillance is necessary to determine whether genotype III spillover from West Texas into metropolitan areas is occurring, potentially initiating urban transmission cycles that could drive SLEV outbreaks.
Our data indicate that SLEV genotype III is established in Texas and importation events continue in the western counties. Despite genotype III circulation in Texas for nearly a decade, human cases have not increased. Since 2002, SLEV is a reportable disease in the state, where case counts are collected by the state contingent on diagnoses. The lack of cases in the northern and western counties could be attributable to several factors, such as the lack of clinical awareness to distinguish SLEV from WNV, the lack of incentive to test for arboviruses, potential misdiagnosis as WNV because of assay cross-reactivity, or the inability of SLEV genotype III from the rural transmission cycle to infect a large enough human population that it would be detected (43,44). Although genotype IIB remains the dominant strain in the Gulf Coast region >40 years after its introduction, whether genotype III possibly was introduced but failed to establish is unclear, given the limitations of sampling and testing in our retrospective study. Ongoing surveillance is critical to ensure that genotype III–associated outbreaks do not go undetected in affected counties and to monitor whether this genotype spreads to the Gulf Coast where an urban cycle with Cx. quinquefasciatus mosquitoes could cause epidemics. Genotype III became the dominant genotype in the western United States in <5 years, raising important questions about why a similar shift did not occur throughout Texas after nearly a decade. Whether any possible geographic and ecologic factors may be preventing the establishment of other genotypes in the Gulf Coast region is unknown. Incorporating accurate clinical data with robust mosquito surveillance data would provide a clearer measure of surveillance coverage across the state, which is currently unknown. Collectively, this study demonstrates the establishment and persistence of SLEV genotype III in Texas, which co-circulates with WNV and can cause concurrent outbreaks, underscoring the need for heightened clinical vigilance and ongoing surveillance.
Dr. Kneubehl is an assistant professor in the Division of Tropical Medicine, Department of Pediatrics, Baylor College of Medicine, Houston, Texas. His research focuses on vector biology and vectorborne diseases with broad interests in genomics, epidemiology, and molecular pathogenesis.
Acknowledgments
The authors express sincere gratitude to the City of Lubbock Health Department and the Zoonoses Control Veterinarian, Public Health Region 1, Texas Department of State Health Services, for their assistance in mosquito collection and processing.
SLEV isolates were obtained through BEI Resources at the National Institutes of Health National Institute of Allergy and Infectious Diseases, as part of the World Reference Center for Emerging Viruses and Arboviruses program: SLEV V07457, NR-49781; SLEV V08449, NR-49782; SLEV V08458, NR-49783; and SLEV, TXAR 9-6038, NR-50061.
This work was funded by intramural startup funds provided to S.E.R. from Texas Children’s Hospital and Baylor College of Medicine Department of Pediatrics. This work also was supported by the Texas Children’s Hospital William T. Schearer Center for Human Immunobiology.
References
- Diaz A, Coffey LL, Burkett-Cadena N, Day JF. Reemergence of St. Louis Encephalitis Virus in the Americas. Emerg Infect Dis. 2018;24:2150–7. DOIPubMedGoogle Scholar
- Reisen WK, Fang Y, Martinez VM. Avian host and mosquito (Diptera: Culicidae) vector competence determine the efficiency of West Nile and St. Louis encephalitis virus transmission. J Med Entomol. 2005;42:367–75. DOIPubMedGoogle Scholar
- Reisen WK, Chiles RE, Martinez VM, Fang Y, Green EN. Experimental infection of California birds with western equine encephalomyelitis and St. Louis encephalitis viruses. J Med Entomol. 2003;40:968–82. DOIPubMedGoogle Scholar
- Gruwell JA, Fogarty CL, Bennett SG, Challet GL, Vanderpool KS, Jozan M, et al. Role of peridomestic birds in the transmission of St. Louis encephalitis virus in southern California. J Wildl Dis. 2000;36:13–34. DOIPubMedGoogle Scholar
- McLean RG, Mullenix J, Kerschner J, Hamm J. The house sparrow (Passer domesticus) as a sentinel for St. Louis encephalitis virus. Am J Trop Med Hyg. 1983;32:1120–9. DOIPubMedGoogle Scholar
- Leake J, Musson E, Chope H. Epidemiology of epidemic encephalitis, St. Louis type. JAMA. 1934;103:728–31. DOIGoogle Scholar
- Curren EJ, Lindsey NP, Fischer M, Hills SLSS. Louis encephalitis virus disease in the United States, 2003–2017. Am J Trop Med Hyg. 2018;99:1074–9. DOIPubMedGoogle Scholar
- Danforth ME, Snyder RE, Feiszli T, Bullick T, Messenger S, Hanson C, et al. Epidemiologic and environmental characterization of the re-emergence of St. Louis encephalitis virus in California, 2015–2020. PLoS Negl Trop Dis. 2022;16:
e0010664 . DOIPubMedGoogle Scholar - Venkat H, Krow-Lucal E, Kretschmer M, Sylvester T, Levy C, Adams L, et al. Comparison of characteristics of patients with West Nile Virus or St. Louis encephalitis virus neuroinvasive disease during concurrent outbreaks, Maricopa County, Arizona, 2015. Vector Borne Zoonotic Dis. 2020;20:624–9. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention (CDC). Outbreak of West Nile-like viral encephalitis—New York, 1999. MMWR Morb Mortal Wkly Rep. 1999;48:845–9.PubMedGoogle Scholar
- Reisen WK, Lothrop HD, Wheeler SS, Kennsington M, Gutierrez A, Fang Y, et al. Persistent West Nile virus transmission and the apparent displacement St. Louis encephalitis virus in southeastern California, 2003-2006. J Med Entomol. 2008;45:494–508.PubMedGoogle Scholar
- Reimann CA, Hayes EB, DiGuiseppi C, Hoffman R, Lehman JA, Lindsey NP, et al. Epidemiology of neuroinvasive arboviral disease in the United States, 1999-2007. Am J Trop Med Hyg. 2008;79:974–9. DOIPubMedGoogle Scholar
- Mackenzie JS, Barrett AD, Deubel V. The Japanese encephalitis serological group of flaviviruses: a brief introduction to the group. Curr Top Microbiol Immunol. 2002;267:1–10. DOIPubMedGoogle Scholar
- Fang Y, Reisen WK. Previous infection with West Nile or St. Louis encephalitis viruses provides cross protection during reinfection in house finches. Am J Trop Med Hyg. 2006;75:480–5. DOIPubMedGoogle Scholar
- Bosco-Lauth AM, Kooi K, Hawks SA, Duggal NK. Cross-protection between West Nile virus and emerging flaviviruses in wild birds. Am J Trop Med Hyg. 2024;112:657–62. DOIPubMedGoogle Scholar
- Kramer LD, Chandler LJ. Phylogenetic analysis of the envelope gene of St. Louis encephalitis virus. Arch Virol. 2001;146:2341–55. DOIPubMedGoogle Scholar
- Rodrigues SG, Nunes MR, Casseb SM, Prazeres AS, Rodrigues DS, Silva MO, et al. Molecular epidemiology of Saint Louis encephalitis virus in the Brazilian Amazon: genetic divergence and dispersal. J Gen Virol. 2010;91:2420–7. DOIPubMedGoogle Scholar
- Diaz LA, Ré V, Almirón WR, Farías A, Vázquez A, Sanchez-Seco MP, et al. Genotype III Saint Louis encephalitis virus outbreak, Argentina, 2005. Emerg Infect Dis. 2006;12:1752–4. DOIPubMedGoogle Scholar
- Venkat H, Krow-Lucal E, Hennessey M, Jones J, Adams L, Fischer M, et al. Concurrent outbreaks of St. Louis encephalitis virus and West Nile Virus disease—Arizona, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1349–50.PubMedGoogle Scholar
- White GS, Symmes K, Sun P, Fang Y, Garcia S, Steiner C, et al. Reemergence of St. Louis encephalitis Virus, California, 2015. Emerg Infect Dis. 2016;22:2185–8. DOIPubMedGoogle Scholar
- Ridenour CL, Cocking J, Poidmore S, Erickson D, Brock B, Valentine M, et al. St. Louis encephalitis virus in the southwestern United States: a phylogeographic case for a multi-variant introduction event. Front Genet. 2021;12:
667895 . DOIPubMedGoogle Scholar - Swetnam DM, Stuart JB, Young K, Maharaj PD, Fang Y, Garcia S, et al. Movement of St. Louis encephalitis virus in the Western United States, 2014- 2018. PLoS Negl Trop Dis. 2020;14:
e0008343 . DOIPubMedGoogle Scholar - Chandler LJ, Parsons R, Randle Y. Multiple genotypes of St. Louis encephalitis virus (Flaviviridae: Flavivirus) circulate in Harris County, Texas. Am J Trop Med Hyg. 2001;64:12–9. DOIPubMedGoogle Scholar
- Lillibridge KM, Parsons R, Randle Y, Travassos da Rosa AP, Guzman H, Siirin M, et al. The 2002 introduction of West Nile virus into Harris County, Texas, an area historically endemic for St. Louis encephalitis. Am J Trop Med Hyg. 2004;70:676–81. DOIPubMedGoogle Scholar
- Williams KH, Hollinger FB, Metzger WR, Hopkins CC, Chamberlain RW. The epidemiology of St. Louis encephalitis in Corpus Christi, Texas, 1966. Am J Epidemiol. 1975;102:16–24. DOIPubMedGoogle Scholar
- Luby JP, Miller G, Gardner P, Pigford CA, Henderson BE, Eddins D. The epidemiology of St. Louis encephalitis in Houston, Texas, 1964. Am J Epidemiol. 1967;86:584–97. DOIPubMedGoogle Scholar
- Kneubehl AR. Texas-SLEV-genome-surveillance. 2025 [cited 2025 Jan 1]. https://github.com/kneubehl/Texas-SLEV-Genome-Surveillance
- Lanciotti RS, Kerst AJ. Nucleic acid sequence-based amplification assays for rapid detection of West Nile and St. Louis encephalitis viruses. J Clin Microbiol. 2001;39:4506–13. DOIPubMedGoogle Scholar
- Wang MX, Lou EG, Sapoval N, Kim E, Kalvapalle P, Kille B, et al. Olivar: towards automated variant aware primer design for multiplex tiled amplicon sequencing of pathogens. Nat Commun. 2024;15:6306. DOIPubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: Improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–22. DOIPubMedGoogle Scholar
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. DOIPubMedGoogle Scholar
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2:
vew007 . DOIPubMedGoogle Scholar - Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- May FJ, Li L, Zhang S, Guzman H, Beasley DWC, Tesh RB, et al. Genetic variation of St. Louis encephalitis virus. J Gen Virol. 2008;89:1901–10. DOIPubMedGoogle Scholar
- Auguste AJ, Pybus OG, Carrington CV. Evolution and dispersal of St. Louis encephalitis virus in the Americas. Infect Genet Evol. 2009;9:709–15. DOIPubMedGoogle Scholar
- Lincoln FC. The waterfowl flyways of North America. Washington: US Department of Agriculture; 1935. p. 7–8.
- Tasi TF, Smith GC, Ndukwu M, Jakob WL, Happ CM, Kirk LJ, et al. Entomologic studies after a St. Louis encephalitis epidemic in Grand Junction, Colorado. Am J Epidemiol. 1988;128:285–97. DOIPubMedGoogle Scholar
- Hayes RO, LaMotte LC, Holden P. Ecology of arboviruses in Hale County, Texas, during 1965. Am J Trop Med Hyg. 1967;16:675–87.PubMedGoogle Scholar
- Gorris ME, Bartlow AW, Temple SD, Romero-Alvarez D, Shutt DP, Fair JM, et al. Updated distribution maps of predominant Culex mosquitoes across the Americas. Parasit Vectors. 2021;14:547. DOIPubMedGoogle Scholar
- Luby JP, Haley RW. Recurrent St. Louis encephalitis infection in residents of a flood plain of the Trinity River, Roosevelt Heights (Dallas, Texas). Am J Epidemiol. 1972;96:107–13. DOIPubMedGoogle Scholar
- Wiseman J, Grimes J, Eads RS. Louis encephalitis outbreak in Cameron County, Texas. Mosq News. 1959;19. DOIGoogle Scholar
- Erickson TA, Ronca SE, Gunter SM, Brown EL, Hasbun R, Murray KO. Zoonotic disease testing practices in pediatric patients with meningitis and encephalitis in a subtropical region. Pathogens. 2022;11:501. DOIPubMedGoogle Scholar
- Vanichanan J, Salazar L, Wootton SH, Aguilera E, Garcia MN, Murray KO, et al. Use of testing for West Nile virus and other arboviruses. Emerg Infect Dis. 2016;22:1587–93. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: March 31, 2026
Table of Contents – Volume 32, Number 4—April 2026
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Shannon E. Ronca, Baylor College of Medicine, Houston, 1102 Bates Ave, Ste 340, Houston, TX 77030, USA
Top