Geographically Distinct Circulation of Genotype II and III St. Louis Encephalitis Virus, Texas, USA, 2009–2024
Alexander R. Kneubehl, Daniel P. Rehm, Michael W. Curtis, Bianca M. Wimmer, Bethany Bolling, Angie Broussard, Jeremy Vela, Jennifer Rocha, Lindsey Templeton, Maximea Vigilant, Courtney Standlee, Steven M. Presley, Job E. Lopez, and Shannon E. Ronca

Author affiliation: Baylor College of Medicine, Houston, Texas, USA (A.R. Kneubehl, D.P. Rehm, M.W. Curtis, J.E. Lopez, S.E. Ronca); Texas Children’s Hospital William T. Schearer Center for Human Immunobiology, Houston (A.R. Kneubehl, D.P. Rehm, S.E. Ronca); Texas Tech University Institute of Environmental and Human Health, Lubbock, Texas, USA (B.M. Wimmer, S.M. Presley); Texas Department of State Health Services, Austin, Texas, USA (B. Bolling); Harris County Public Health Division of Mosquito and Vector Control, Houston (A. Broussard, J. Vela, J. Rocha, L. Templeton, M. Vigilant, C. Standlee)
Main Article
Figure 5
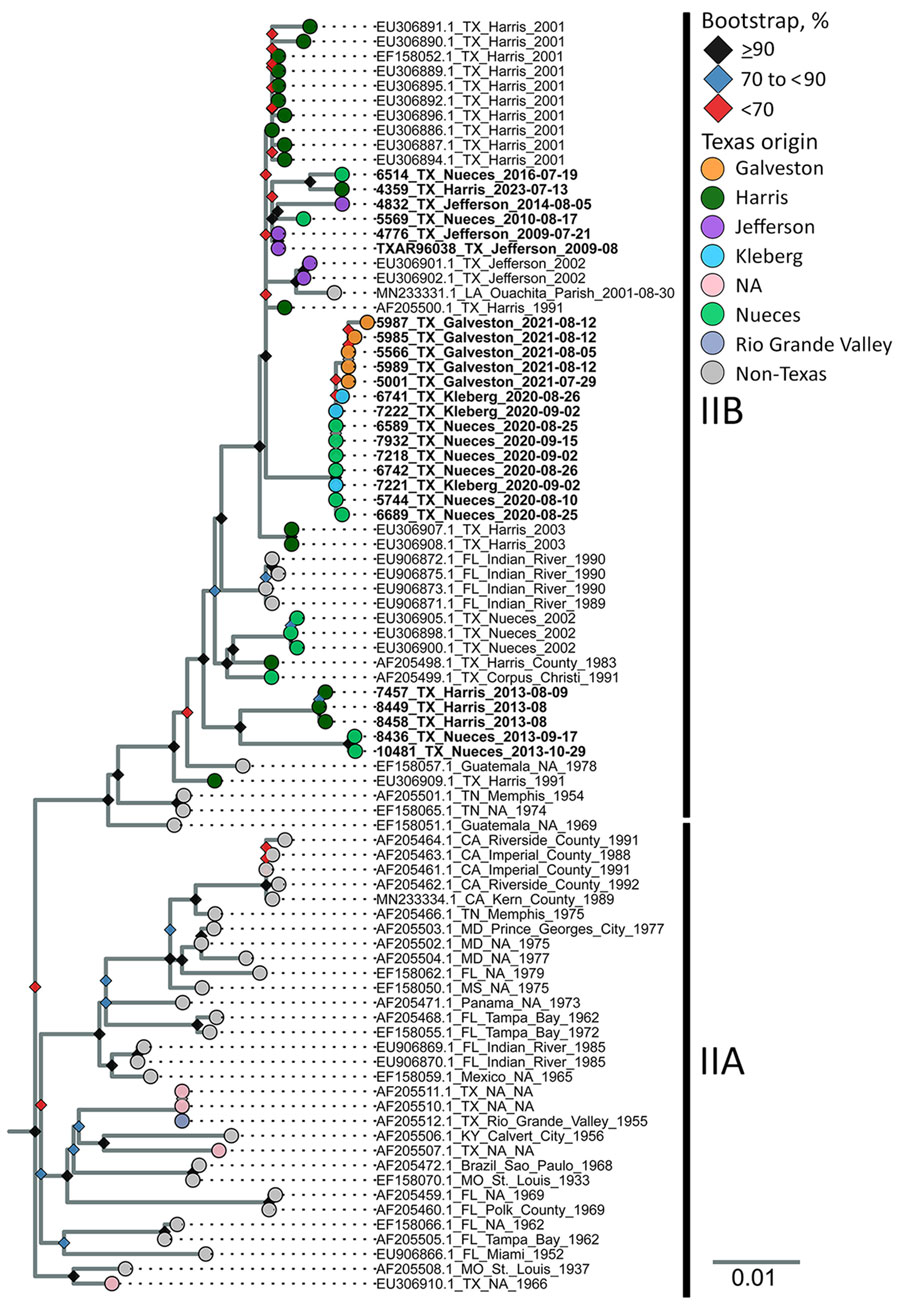
Figure 5. Maximum-likelihood phylogenic analysis of St. Louis encephalitis virus genotype IIA and IIB envelope gene sequences from study of circulation of genotype II and III St. Louis encephalitis virus, Texas, USA. Maximum-likelihood inferred tree of 86 SLEV genotype II envelope gene nucleotide sequences. Two genotype III outgroup sequences (not shown) were used to root tree (GenBank accession nos. FJ753287.2 and FJ753286.2). Tip labels indicate the GenBank accession number, sample origin, and collection date. Bold tip labels indicate samples generated by this study. Tip colors highlight different sample origins from within Texas, by county (or area, in the case of the Rio Grande Valley sample). Bootstrap support is shown at nodes; different colors indicate level of bootstrap support from 10,000 ultrafast bootstrap replicates. Scale bar indicates number of nucleotide substitutions per site. NA, not available (missing or unknown).
Main Article
Page created: March 04, 2026
Page updated: April 15, 2026
Page reviewed: April 15, 2026
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.