Volume 6, Number 3—June 2000
Research
Rhinosporidium seeberi: A Human Pathogen from a Novel Group of Aquatic Protistan Parasites
Cite This Article
Citation for Media
Abstract
Rhinosporidium seeberi, a microorganism that can infect the mucosal surfaces of humans and animals, has been classified as a fungus on the basis of morphologic and histochemical characteristics. Using consensus polymerase chain reaction (PCR), we amplified a portion of the R. seeberi 18S rRNA gene directly from infected tissue. Analysis of the aligned sequence and inference of phylogenetic relationships showed that R. seeberi is a protist from a novel clade of parasites that infect fish and amphibians. Fluorescence in situ hybridization and R. seeberi-specific PCR showed that this unique 18S rRNA sequence is also present in other tissues infected with R. seeberi. Our data support the R. seeberi phylogeny recently suggested by another group. R. seeberi is not a classic fungus, but rather the first known human pathogen from the DRIPs clade, a novel clade of aquatic protistan parasites (Ichthyosporea).
Figure 1

Figure 1. Histology of rhinosporidiosis. A formaldehyde-fixed section of human nasal polyp was stained with Periodic acid-Schiff (PAS) and visualized by bright-field microscopy at 400X magnification. The thick walls of immature R. seeberi...
Rhinosporidiosis manifests as slow-growing, tumorlike masses, usually of the nasal mucosa or ocular conjunctivae of humans and animals. Patients with nasal involvement often have unilateral nasal obstruction or bleeding due to polyp formation. The diagnosis is established by observing the characteristic appearance of the organism in tissue biopsies (Figure 1). Treatment consists of surgical excision, but relapse occurs in approximately 10% of patients (1); antimicrobial therapy is not effective (2). Rhinosporidiosis occurs in the Americas, Europe, Africa, and Asia but is most common in the tropics, with the highest prevalence in southern India and Sri Lanka. A survey of schoolchildren from Pallam, India, found 11 cases in 781 children examined (prevalence 1.4%) (3). Autochthonous cases have been reported from the southeastern United States (4). Studies have linked infection to swimming or bathing in freshwater ponds, lakes, or rivers (2,5).
The etiologic agent of rhinosporidiosis, Rhinosporidium seeberi, is an enigmatic microbe that has been difficult to classify. Recently, R. seeberi has been considered a fungus, but it was originally thought to be a protozoan parasite (2). Its morphologic characteristics resemble those of Coccidioides immitis: both organisms have mature stages that consist of large, thick-walled, spherical structures containing smaller daughter cells (endospores). In addition, R. seeberi is visualized with fungal stains such as methenamine silver and Periodic acid-Schiff, as well as mucicarmine, which stains the fungus Cryptococcus neoformans. R. seeberi has not been detected in the environment, and its natural host or reservoir is unknown. Attempts to propagate this organism on artificial media have failed, as has continuous cocultivation with human cell lines (6).
We report a molecular approach for establishing the phylogenetic relationships R. seeberi to other eukaryotes. This approach is based on amplification of the small subunit rRNA gene sequence from infected tissue, as in the method used to identify the culture-resistant bacillus of Whipple disease (7). The sequence of the small subunit rRNA gene has proven to be a useful gauge of evolutionary relationships for many organisms from diverse taxonomic groups (8).
Specimens
We obtained a sample of frozen, minced, infected canine nasal polyp in tissue culture media that had been used for the limited propagation of R. seeberi in cell culture (6). After thawing and a 1-minute centrifugation at 500 x g, approximately 0.2 g of the tissue pellet was digested by mechanical disruption and the DNA was purified by adsorption to glass milk in the presence of a chaotropic agent, according to the manufacturer's instructions (Fast Prep, Bio 101, Vista, CA). The DNA was resuspended in 100µL of 10 mM Tris, 1 mM EDTA buffer at pH 8.5.
Three blocks of fixed, paraffin-embedded nasal polyps from unrelated patients with histologically confirmed rhinosporidiosis were obtained. One tissue sample came from a patient born in southern Asia but living in the United States, and two samples came from patients living in Spain. Twenty-three blocks of fixed, paraffin-embedded nasal polyps from patients without rhinosporidiosis were obtained from 12 consecutive patients at the Palo Alto Veterans Affairs (VA) hospital who had undergone nasal polypectomy. Two 25-µm sections were cut from each block, and the sections were deparaffinized and digested (5). DNA from the digests was then purified by the Isoquick method (ORCA Research, Inc., Bothell, WA), and the DNA was resuspended in 25µL of Tris/EDTA buffer. Sections from a block of fixed, paraffin-embedded human lymph node (histologically normal) were also used in some experiments as negative controls. Tissue sections of C. immitis in bone were obtained from a patient with disseminated coccidioidomycosis at the Palo Alto VA hospital and used for fluorescence in situ hybridization (FISH). Samples of Rosette agent and Dermocystidium salmonis DNA were obtained from the Bodega Marine Laboratory of the University of California, Davis.
Fungal specimens in culture were obtained from laboratories at Stanford University and the Palo Alto VA Health Care System. A cotton swab was used to transfer fungal cells from agar into a 1.5-mL microfuge tube containing 0.5 mL digestion buffer (9) and 0.1 mL glass beads. Samples were incubated at 55°C overnight, and the proteinase k was inactivated at 95°C for 10 minutes and then subjected to two freeze-thaw cycles by immersing tubes in a dry ice-isopropranol bath followed by vortex mixing.
Consensus Polymerase Chain Reaction (PCR) of the 18S rRNA Gene
Broad-range fungal PCR primers were designed from a database of >4,000 small subunit rDNA sequences, with the ARB software package (Technical University, Munich, Germany). Primers were selected that would anneal to most fungal and some protist 18S rDNA but not to 18S rDNA from the chordata (F1-fw, F2-rev, F3-rev) (Table). When the specificity of the primer pairs was tested by using human lymphocyte DNA, no amplification was observed (data not shown). The broad range of primers F1-fw/F2-rev was tested by using DNA from several diverse fungi. Amplification products of the expected size were produced by using DNA from Aspergillus oryzae, Alternaria alternata, Candida albicans, Saccharomyces cerevisiae, Trichyphyton rubrum, Panus rudis, Neurospora crassa, Fusarium solani, Beauveria bassiana, Flammulina velutipes, Gibberella zeae, and Pleurotus ostreatus (data not shown).
PCR consisted of 40 cycles of amplification on a Perkin-Elmer GeneAmp 2400 thermal cycler. After an initial activation of Taq gold at 94°C for 10 minutes, each cycle consisted of 30 seconds of melting at 94°C, 30 seconds of annealing at 56°C, and 30 seconds of extension at 72°C. The last cycle was followed by an extension step at 72°C for 7 minutes. Amplification products were detected by electrophoresis on 2% agarose gels stained with ethidium bromide and visualized with a UV transilluminator.
On the basis of the sequences obtained by consensus PCR with primers F1-fw/F2-rev (~500 bp) and F1-fw/F3-rev (~1000 bp), primers Dermo-fw and Dermo-rev were designed (Table) and used in a PCR to amplify a more complete 18S rDNA sequence of R. seeberi.
Rhinosporidium-Specific PCR
A pair of PCR primers was designed from unique regions of the R. seeberi 18S rRNA gene sequence (Rhino-fw and Rhino-rev) (Table). These primers were used in a 50-µL PCR as described, except that AmpliTaq DNA polymerase (PE-ABI) was used at 1 unit per reaction, no dimethyl sulfoxide was added, 50 cycles of PCR were run with a 3-minute pre-melt at 94°C, and the annealing temperature was 55°C. To each 50-µL PCR reaction, 1µL or 5µL of purified DNA were added.
ß-Globin PCR
ß-globin PCR was performed on control tissues as described previously (9).
DNA Cloning, Sequencing, and Phylogenetic Analysis
Amplification products were cloned by using the Topo-TA cloning kit (Invitrogen, Carlsbad, CA), and three clones were sequenced. Each clone consisted of 1,750 bp of 18S rDNA. Priming sequences were removed for further analysis, yielding 1,699 bp of meaningful sequence. DNA sequencing was performed as described (10). A consensus sequence from the three clones was made to correct for any Taq polymerase incorporation errors. The 18S rDNA primers (Table) were used as sequencing primers.
The R. seeberi 18S rDNA sequence was aligned by using the automated aligner of the ARB software package. Ambiguously and incorrectly aligned positions were manually aligned on the basis of the conserved primary sequence and secondary structure. The phylogenetic relationship of R. seeberi to other eukaryotes was inferred from 1,350 unambiguously aligned (masked) positions with a maximum-likelihood algorithm (11,12), on the basis of a previously aligned dataset of the DRIPs clade (named after the organisms Dermocystidium, the Rosette agent, Ichthyophonus, and Psorospermium) (13). The dataset was used to empirically determine nucleotide frequencies and instantaneous substitution rates with the restriction of a 2:1 transition to transversion ratio. The organisms used in our tree and the accession numbers for their small subunit rRNA sequences include Artemia salina (X01723), Xenopus laevis (X04025), Mytilus edulis (L24489), Tripedalia cystophora (L10829), Microciona prolifera (L10825), Diaphanoeca grandis (L10824), Rosette agent (L29455), R. seeberi (AF158369), Dermocystidium species (U21336), Dermocystidium salmonis (U21337), Psorospermium haeckelii (U33180), Ichthyophonus hoferi (D14358), Aspergillus fumagatus (M60300), Chytridium confervae (M59758), Mucor racemosus (X54863), Acanthamoeba castellanii (U07413), Zamia pumila (M20017), Porphyra spiralis (L26177), Lagenidium giganteum (X54266), Labyrinthuloides minuta (L27634), Perkinsus marinus (X75762), Sarcocystis muris (M64244). The tree topology was confirmed by using a neighbor-joining algorithm with Jukes-Cantor corrected distance values and a maximum-parsimony algorithm (ARB). The nucleotide sequence for the partial 18S rRNA gene of R. seeberi has been deposited in GenBank (accession number AF158369).
Fluorescence in Situ Hybridization (FISH)
Tissue sections on slides were dewaxed by immersion in 99% octane (Sigma, St. Louis, MO). Samples subjected to FISH included R. seeberi-infected human nasal polyps, C. immitis-infected bone, a Rosette agent-infected cell line, and smears of C. albicans. The Rhinosporidium probe was based on the Rhino-rev 18S rDNA primer and was biotinylated at both the 5' and 3' ends (Table). The control probe, which consisted of the complement of the Rhinosporidium probe, was also biotinylated at both ends. To each slide, 50 ng of biotinylated probe in 30 µL of hybridization buffer was added. Cover slips were placed, and the slides were incubated at 40°C overnight in a humid chamber. The hybridization buffer consisted of 10% dextran, 0.2% bovine serum albumin, and 0.01% polyadenosine, in 5X SET buffer; the 25X SET buffer consisted of 3.75M sodium chloride, 25 mM EDTA, and 0.5M Tris at pH 7.8. Cover slips were removed by immersion in 5X SET buffer at 4°C, and the slides were washed for 10 minutes per cycle, twice in 0.2X SET buffer at 25°C and once at 40°C. The slides were then subjected to tyramide signal amplification according to the manufacturer's instructions (TSA indirect, NEN Life Sciences, Boston, MA). Cy5-streptavidin (Amersham, Piscataway, NJ) at 1 mg/mL was diluted 1:500 and added to the slides for fluorescence signal detection. Tissue sections were visualized on a Bio Rad confocal microscope at 200X magnification after the application of 15-20µL of Vectashield mountant (Sigma) and a cover slip.
Electron Microscopy
A portion of formalin-fixed, paraffin-embedded nasal polyp from a patient with rhinosporidiosis was removed from the block, dewaxed with xylene, rehydrated with ethanol, post-stained with 1.5% osmium tetroxide, then dehydrated with ethanol, transferred to propylene oxide followed by Epon 12 resin, heat-catalyzed at 65°C, and ultrasectioned at 50 nm. The grid-mounted sections were then serially stained with lead hydroxide and uranyl acetate and examined with a Phillips 201 electron microscope at 75KV.
Phylogenetic Classification of R. seeberi Inferred from the 18S rRNA Gene
Figure 2
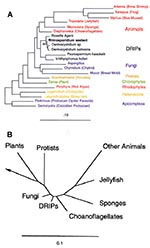
Figure 2. Phylogeny of Rhinosporidium seeberi and the DRIPs clade of protists (Ichthyosporea). A. Phylogenetic tree inferred from the 18S rDNA sequences of R. seeberi and other selected eukaryotes by using a maximum...
Consensus PCR of the 18S rRNA gene with DNA from a digest of an R. seeberi-infected canine nasal polyp produced amplification products of the expected size visible on gel electrophoresis (primer pairs F1-fw/F2-rev = ~500 bp and F1-fw/F3-rev = ~1000 bp) (data not shown). No amplification product was detected by using control tissue and reagents. On the basis of the initial phylogenetic assessment of these sequences, primers Dermo-fw and Dermo-rev were designed for amplification of a more complete portion of the R. seeberi 18S rRNA gene. Our phylogenetic analysis of this gene suggests that R. seeberi is a member of the DRIPs clade of aquatic protistan parasites (Figure 2). The nearest evolutionary neighbors of R. seeberi for which a sequence is available are members of the Dermocystidium genus, which infect salmon and trout.
Development and Use of a Rhinosporidium-Specific PCR Assay
A PCR assay specific for R. seeberi was developed. Primers were created by aligning 18S rDNA sequences from R. seeberi, members of the DRIPs clade, Saccharomyces cerevisiae, and humans. The Rhinosporidium primers (Rhino-fw, Rhino-rev) each have three nucleotide mismatches with the sequences from the nearest phylogenetic relatives in the Dermocystidium genus and multiple other mismatches with fungal and human 18S rDNA sequences. An assay sensitivity of 1-10 gene copies was demonstrated by using a dilution series of cloned R. seeberi 18S rDNA. The specificity of the assay was assessed with DNA from human lymphocytes, S. cerevisiae, D. salmonis, and the Rosette agent. No amplification was detected when these DNA samples were used in the specific PCR assay, although product was amplified from these samples with either ß-globin primers (lymphocytes) or broad-range 18S rDNA primers (F1-fw/F2-rev) (Table).
Figure 3

Figure 3. Agarose gel electrophoresis of Rhinosporidium-specific PCR products. The specific amplification product is 377 bp. No amplification product is seen in the negative control samples consisting of water (reagent-only control), digestion buffer...
The original DNA sample from the infected canine nasal polyp yielded a product of the expected size with the Rhinosporidium-specific PCR assay (Figure 3). Purified DNA from the tissue blocks of human nasal polyps resected from three patients with rhinosporidiosis also yielded positive results in this PCR assay, with visible bands seen on gel electrophoresis (RS 1-3) (Figure 3). Direct sequencing of these PCR products demonstrated complete identity over the 377 bp with the cloned sequence from the canine polyp. DNA samples from 23 nasal polyp specimens resected from 12 patients without rhinosporidiosis were subjected to both Rhinosporidium-specific and ß-globin PCR assays. These uninfected polyps failed to yield visible amplicons after gel electrophoresis of the Rhinosporidium-specific PCR reactions (data not shown). However, all these samples yielded visible amplification products after ß-globin PCR, demonstrating that amplifiable DNA was present, without substantial PCR inhibition. These results confirmed that the presence of the putative R. seeberi 18S rDNA sequence correlated with the presence of disease (rhinosporidiosis).
Fluorescence in Situ Hybridization
Figure 4

Figure 4. An oligonucleotide probe complementary to a unique region of the Rhinosporidium seeberi 18S rRNA sequence localizes to visible organisms in human nasal tissue by using fluorescence in situ hybridization. Confocal micrographs...
We sought to determine by using FISH if the R. seeberi 18S rDNA sequence was linked to visible pathology in tissue. The Rhinosporidium 18S rRNA probe but not the control probe bound to R. seeberi organisms in tissue, providing further evidence that the putative R. seeberi 18S rRNA sequence is present in R. seeberi organisms (Figure 4). To test the specificity of the hybridization, smears of the fungus C. albicans, cells infected with the Rosette agent, and tissue sections of C. immitis were subjected to FISH. No specific hybridization to these organisms was detected by using the Rhinosporidium 18S rRNA probe compared with the control probe (data not shown).
Electron Microscopy
Figure 5

Figure 5. Transmission electron micrograph of a mitochondrion from Rhinosporidium seeberi. The cristae of this mitochondrion (arrows) have tubulovesicular morphology. Magnification 195,000X.
Transmission electron micrographs were taken of R. seeberi trophocytes in a human nasal polyp. Multiple mitochondria were visualized, showing that the cristae had tubulovesicular morphology (Figure 5), unlike the flat cristae found in fungi.
In the 1890s, first Malbran and then Seeber (14) described an apparent sporozoan parasite in nasal polyps from patients living in Argentina. Seeber's teacher, Wernicke, named the organism Coccidium seeberia after the protozoal subdivision Coccidia and his pupil, Guillermo Seeber (2). In 1923, Ashworth described the life cycle of the organism, argued that it is a fungus, and proposed the name R. seeberi (15). Since then, the microbe has been considered a fungus by most microbiologists, although its taxonomy has been debated (1,2,16). Using a consensus PCR approach, we amplified a unique 18S rDNA sequence from a canine nasal polyp infected with R. seeberi. To prove that this unique 18S rDNA sequence came from R. seeberi, we sought to fulfill sequence-based guidelines for microbial disease causation, since Koch's postulates cannot be fulfilled for uncultivated microbes (17). Using a Rhinosporidium-specific PCR assay, we showed that the unique rDNA sequence present in the canine polyp was also present in three human polyp samples from patients with rhinosporidiosis. We showed the specificity of this association by demonstrating that this rDNA sequence was not present in control nasal polyps from 12 patients without rhinosporidiosis. We also used FISH to link the putative R. seeberi 18S rRNA sequence to organisms visible in tissue. Although the Rhinosporidium probe localized to R. seeberi organisms in tissue using FISH, the probe did not localize to two fungal organisms or another member of the DRIPs clade (Rosette agent), suggesting some specificity to the hybridization.
Sequence-based data also provide support for a causal relationship when a microbial genotype (e.g., phylogenetic placement) correctly predicts microbial phenotype and host response (17). In other words, the nature of the pathogen inferred from phylogenetic analysis of its nucleic acid sequence should be consistent with the known biologic characteristics of closely related microbes and with the nature of the disease. Therefore, the nearest phylogenetic neighbors to R. seeberi could be predicted to have similarities in morphology, tissue histology, and pathogenesis. D. salmonis, the closest known relative to R. seeberi, has a large spherical structure containing endospore-like daughter cells (18). Histology of infected hosts shows gill inflammation and epithelial hyperplasia. The resemblance between R. seeberi and fish pathogens has been noted before. In 1960, Satyanarayana wrote in his review of 255 cases of rhinosporidiosis (1) that since R. seeberi has a morphology similar to those of some fish parasites, it "..may also be a parasite or saprophyte of fish and that man, equines, and cattle obtain the infection through water in which fish harbouring the parasite live."
A recent independent report based on amplification of 18S rDNA from two human rhinosporidiosis tissue samples also concludes that R. seeberi is a member of the DRIPs clade of microbes (19). Our data support this conclusion and provide more evidence for a causal relationship. In addition, we describe a Rhinosporidium-specific PCR assay that can be used for detecting this organism in clinical and environmental samples. Our 18S rDNA sequence differs from the sequence determined by these investigators (GenBank AF118851) at a single position out of 1,699 common bases. We excluded from analysis sequence derived from our PCR primers, as amplicons will contain the primer sequences regardless of the target sequence as long as there is partial annealing during PCR, leading to potentially spurious conclusions about sequence in these regions. The 18S rDNA sequence of R. seeberi determined by this group includes the primer sequences.
Although we describe early trophocytes with mitochondrial cristae having vesicular ultrastructure, other investigators have found sporangia with mitochondrial cristae having a flat ultrastructure (19). The different mitochondrial morphologies observed may be due to differences in developmental stage of the organism or in methods of tissue preparation for electron microscopy. Nevertheless, another member of the DRIPs clade, Ichthyophonus hoferi, has vesicular mitochondrial cristae. Classic fungi (Eumycota) have flat mitochondrial cristae.
Knowledge of the molecular phylogeny of R. seeberi is more than an exercise in taxonomy. For organisms such as R. seeberi, which are difficult to grow in the laboratory, phylogenetic analysis provides some insight into the characteristics of the organism that can be used to further our understanding of disease pathogenesis and epidemiology, as well as to improve diagnosis and treatment. Knowing that R. seeberi is a member of the DRIPs clade of microbes allows hypotheses to be generated about how it causes human disease by analogy, drawing on the knowledge and experience of the veterinary sciences. The separate but linked observations that rhinosporidiosis in humans is associated with exposure to water and that R. seeberi belongs to a clade of aquatic parasites lead to a testable hypothesis: the natural hosts of R. seeberi are fish or other aquatic animals, and humans acquire infection when they come into contact with water containing these fish and their parasites. Investigators should therefore look for evidence of infection in fish in ponds and rivers in disease-endemic areas. From a public health perspective, the R. seeberi-specific PCR assay can be used to study environmental sources of infection (e.g., specific bodies of water) and may provide a means of preventing disease through identification of infected water.
Conversely, knowing that R. seeberi is a member of the DRIPs clade may help us understand this important, distinct group of microbes that appear to form the deepest branch in the animal lineage. R. seeberi is a member of a newly recognized group of human and animal pathogens; the name Ichthyosporea has been proposed for this expanding taxon of microbes (20). Little information is available about these organisms and how they cause disease. We hope that collaborations between researchers in human and animal medicine will correct this deficiency.
Multiple antimicrobials, including antifungal agents, have been used in the treatment of rhinosporidiosis, based on the belief that R. seeberi is a fungus. However, no antimicrobial agent is clearly effective. The medical treatment of rhinosporidiosis may be improved through screening antiparasitic drugs for an effect on disease in Dermocystidium-infected fish or infected cell lines.
In conclusion, phylogenetic analysis of the R. seeberi 18S rRNA gene suggests that this culture-resistant organism is not a member of the Eumycota, but rather is the first known human pathogen from a novel clade of aquatic protistan parasites that form a branch in the evolutionary tree near the animal-fungal divergence. R. seeberi-specific PCR and FISH confirm the association of this unique 18S rDNA sequence with the presence of rhinosporidiosis. This knowledge can be used to further our understanding of the natural reservoir of this organism and the risk factors, pathogenesis, and treatment of this disease. This discovery also expands our appreciation of the diversity among eukaryotic organisms that are pathogenic to humans and highlights the limitations of basing phylogenetic classification on morphology alone.
Dr. Fredricks is a research associate in the Division of Infectious Diseases, Stanford University. He studies the use of nucleic acid sequences to detect and identify microbial pathogens, including those that are novel or uncultivated.
Acknowledgments
We thank Josh Fierer and Jesus Gonzales for providing human Rhinosporidium tissue blocks, Mike Levy for the canine nasal polyp used for consensus PCR, Kristin Arkush for the Dermocystidium salmonis and Rosette agent DNA, and Robin Gutell for the mask used for phylogenetic analysis of the DRIPs clade. Bob Metzenberg, Sara Fulz, Larry Mirels, and the staff of the clinical microbiology laboratories at Stanford and the Palo Alto VA hospitals supplied fungal isolates for testing broad-range 18S rDNA primers.
This study was supported by grants from the National Institutes of Health (K11-AI01360 D.N.F.) and the Donald B. and Delia B. Baxter Foundation (D.A.R).
References
- Satyanarayana C. Rhinosporidiosis with a record of 255 cases. Acta Oto-Laryng. 1960;51:348–66. DOIGoogle Scholar
- Kwon-Chung KJ, Bennett JE. Rhinosporidiosis. In: Medical mycology. Philadelphia: Lea & Febiger; 1992. p. 695-706.
- Moses JS, Shanmugham A. Epidemiological survey of rhinosporidiosis in man--a sample survey in a high school located in a hyperendemic area. Indian Vet J. 1987;64:34–8.
- Gaines JJ, Clay JR, Chandler FW, Powell ME, Sheffield PA, Keller A. Rhinosporidiosis: three domestic cases. South Med J. 1996;89:65–7.PubMedGoogle Scholar
- Kennedy FA, Buggage RR, Ajello L. Rhinosporidiosis: a description of an unprecedented outbreak in captive swans (Cygnus spp.) and a proposal for revision of the ontogenic nomenclature of Rhinosporidium seeberi. J Med Vet Mycol. 1995;33:157–65. DOIPubMedGoogle Scholar
- Levy MG, Meuten DJ, Breitschwerdt EB. Cultivation of Rhinosporidium seeberi in vitro: interaction with epithelial cells. Science. 1986;234:474–6. DOIPubMedGoogle Scholar
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple's disease. N Engl J Med. 1992;327:293–301.PubMedGoogle Scholar
- Pace NR. A molecular view of microbial diversity in the biosphere. Science. 1997;276:734–40. DOIPubMedGoogle Scholar
- Fredricks DN, Relman DA. Paraffin removal from tissue sections for digestion and PCR analysis. Biotechniques. 1999;26:198–200.PubMedGoogle Scholar
- Fredricks DN, Relman DA. Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 1998;36:2810–6.PubMedGoogle Scholar
- Olsen GJ, Matsuda H, Hagstrom R, Overbeek R. fastDNAmL: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput Appl Biosci. 1994;10:41–8.PubMedGoogle Scholar
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–76. DOIPubMedGoogle Scholar
- Ragan MA, Goggin CL, Cawthorn RJ, Cerenius L, Jamieson AV, Plourde SM, A novel clade of protistan parasites near the animal-fungal divergence. Proc Natl Acad Sci U S A. 1996;93:11907–12. DOIPubMedGoogle Scholar
- Seeber G. Un nuevo esporozuario parasito del hombre: dos casos encontrados en polipos nasales. Thesis, Universidad Nacional de Buenos Aires, 1900.
- Ashworth JH. On Rhinosporidium seeberi with special reference to its sporulation and affinities. Trans R Soc Edinb. 1923;53:302–42.
- Kwon-Chung KJ. Phylogenetic spectrum of fungi that are pathogenic to humans. Clin Infect Dis. 1994;19(Suppl 1):S1–7.PubMedGoogle Scholar
- Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch's postulates. Clin Microbiol Rev. 1996;9:18–33.PubMedGoogle Scholar
- Olson RE, Dungan CF, Holt RA. Water-borne transmission of Dermocystidium salmonis in the laboratory. Dis Aquat Organ. 1991;12:41–8. DOIGoogle Scholar
- Herr RA, Ajello L, Taylor JW, Arseculeratne SN, Mendoza L. Phylogenetic analysis of Rhinosporidium seeberi's 18S small-subunit ribosomal DNA groups this pathogen among members of the protoctistan Mesomycetozoa clade. J Clin Microbiol. 1999;37:2750–4.PubMedGoogle Scholar
- Cavalier-Smith T. Neomonada and the origin of animals and fungi. In: Coombs G, Vickerman K, Sleigh M, Warren A, editors. Evolutionary relationships among protozoa. London: Chapman and Hall; 1998. p. 375-407.
Figures
Table
Cite This ArticleTable of Contents – Volume 6, Number 3—June 2000
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
David N. Fredricks, Veterans Affairs, Palo Alto Health Care System, 154-T, 3801 Miranda Ave, Palo Alto, CA 94304, USA; fax: 650-852-3291
Top