Volume 6, Number 5—October 2000
Synopsis
Antigenic Variation in Vector-Borne Pathogens
Cite This Article
Citation for Media
Abstract
Several pathogens of humans and domestic animals depend on hematophagous arthropods to transmit them from one vertebrate reservoir host to another and maintain them in an environment. These pathogens use antigenic variation to prolong their circulation in the blood and thus increase the likelihood of transmission. By convergent evolution, bacterial and protozoal vector-borne pathogens have acquired similar genetic mechanisms for successful antigenic variation. Borrelia spp. and Anaplasma marginale (among bacteria) and African trypanosomes, Plasmodium falciparum, and Babesia bovis (among parasites) are examples of pathogens using these mechanisms. Antigenic variation poses a challenge in the development of vaccines against vector-borne pathogens.
Immunodominant antigens are commonly used to distinguish strains of a species of pathogens. These antigens can vary from strain to strain to the extent that the strain-specific immune responses of vertebrate reservoirs determine the population structure of the pathogen. One such strain-defining antigen is the OspC outer membrane protein of the Lyme disease spirochete Borrelia burgdorferi in the northeastern United States (1). Different strains express different OspC surface proteins in the rodent reservoirs of B. burgdorferi. The single type of ospC gene in a cell does not vary during infections of immunocompetent mammals (2). OspC sequences are diverse, and the immune responses to them appear to provide for balancing selection. This diversity between strains in an immunodominant antigen is often called antigenic variation.
True antigenic variation, however, arises in a single clone or genotype in a single host and "involves the loss, gain, or change in a particular antigenic group, usually by loss, gain, or change in one of the polypeptide or polysaccharide antigens…" (3). In most cases, this change is reversible, i.e., the information for producing the original antigen is archived in the cell and can be used in the future. The adaptive immune system of an infected vertebrate selects against the original infecting serotype, but that specific response is ineffective against new variants. One example of antigenic variation occurs in B. hermsii, a cause of tickborne relapsing fever (4), which has a protein homologous to the OspC protein of B. burgdorferi. However, instead of a single version of this gene, each cell of B. hermsii has several copies of silent genes (alleles) that may be expressed during infection. The sequences of these alleles within a single strain of B. hermsii vary as widely as the ospC alleles of different strains of B. burgdorferi.
We review infectious pathogens that undergo clonal antigenic variation and, like B. hermsii, depend on arthropod vectors for transmission. These pathogens are not free-living and do not form spores or have equivalent means for survival outside an animal. Vertical transmission in the arthropod or the vertebrate either does not occur or is too rare to maintain the pathogen in nature. Without access to another vertebrate host through an arthropod, the pathogen will die with the host.
We restrict this review to situations in which an immune response against an antigen is synonymous with selection for another allele in the population. Many pathogens have repetitive-gene families. A multimember family may resemble a variable antigen gene repertoire in its diversity but may have little or no effect on immunity against infection. An example is the bdr family of genes in B. burgdorferi (6).
Viruses are also said to have antigenic variation but are excluded from this review because the mechanism they use usually depends either on the accumulation of point mutations in a single genotype (e.g., the antigenic drift of influenza A virus) or on recombination or reassortment between two different genotypes infecting the same host (e.g., antigenic shift of influenza A virus). A possible exception is the African swine fever virus, a poxvirus-like linear DNA virus transmitted by soft ticks. These large viruses have tandem repeated genes at their telomeres that undergo deletions during infection (5).
Figure 1
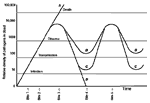
Figure 1. Relative densities of a vector-borne pathogen in the blood of four hosts, A-D. The gray horizontal lines represent the lower thresholds for the persistence of infection, transmission to a new vector,...
Infection with a vector-borne pathogen that undergoes clonal antigenic variation has several possible outcomes (Figure 1). Acquisition of the pathogen by the vector does not in itself constitute transmission. The vector may become infected by a blood meal, but enough pathogens may not be present in the blood for the vector to transmit the infection to its next host. Pathogen peaks may not be so well delineated, especially during late infection when the growth-and-decline curves for individual variants begin to overlap.
Borrelia spp., Anaplasma and related genera, African trypanosomes, Plasmodium spp., and Babesia spp. are vector-borne pathogens that use antigenic variation to evade the host's immunity. The details of antigenic variation differ, but some features are the same; for example, the use of multiphasic antigenic variation or a change among at least three variable antigens rather than alternating between two. At least 10--and sometimes many more--variants or serotypes may be expressed during a single infection. There is a complete or near-complete gene for each of the variable proteins. Variation is achieved by switching one of the several genes expressed at any one time, rather than by accumulating mutations in a single expressed gene, as commonly occurs in viruses. In the best-studied examples, African trypanosomes and relapsing fever Borrelia spp., the rate of antigen switching in the vertebrate host is approximately the same, at 10-4 to 10-2 per cell per generation.
True antigenic variation has been demonstrated in other human pathogens, including Neisseria gonorrhoeae, Mycoplasma spp., Campylobacter fetus, Pneumocystis carinii, and Giardia lamblia. In addition, the complete genomic sequences of other pathogenic bacteria, such as Helicobacter pylori, Treponema pallidum, and Mycobacterium tuberculosis, encompass large families of repeated genes that are polymorphic in sequence and may be involved in antigenic variation.
The vector-borne pathogens, in particular the African trypanosomes and relapsing fever Borrelia spp., offer the least ambiguous models for understanding the biology and evolution of antigenic variation. These pathogens depend on hematophagous arthropods for transmission to new vertebrate hosts, and consequently, the likelihood of transmission is a direct function of the duration and density of the pathogens in the blood. Blood-cell types are comparatively simple, and antibodies alone can clear infection by relapsing fever Borrelia spp. and African trypanosomes (8,9). Apparently these two pathogens need to defend only against humoral immunity.
Figure 2
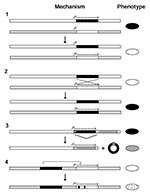
Figure 2. Four genetic mechanisms for antigenic variation in a hypothetical pathogen. 1. Modification of transcript levels. Two loci are shown in the figure for black and white genes. When the black gene...
Vector-borne pathogens use one or more genetic mechanisms to circumvent the immune system. Four general mechanisms for antigenic variation have been described (10): modification of transcript levels, gene conversion, DNA rearrangement, and multiple point mutations (Figure 2). An example of the first mechanism is the reversible activation of expression of a variable antigen gene at one locus as expression of a previously active variable antigen gene at another locus in the genome becomes silent, an event that occurs without DNA changes at the loci themselves. In the figure, pathogen X has surface antigen genes black and white at two loci. At each locus there is a potential promoter, but only at the black locus is a gene expressed. After a switch, the black locus is silent, but the white locus 3 is active.
The second mechanism, gene conversion, is probably the most widespread for replacing expression of one gene with another. The change may be complete, thereby altering all defining epitopes of the antigen, or partial, for example, when a central hypervariable region of a protein is replaced through cross-overs in highly similar flanking regions. This process commonly involves genes on separate chromosomes or plasmids in the cell but can also occur within the same replicon. When one gene is displaced at an expression site, the organism uses for that replacement a copy of a gene from a more stable location in the genome. In Figure 2, a black gene converts a white gene at a site with an active promoter. Gene conversion allows a pathogen to retain a complete repertoire of variable antigen genes.
More extensive change in the genotype may occur by the third mechanism, DNA rearrangement. In Figure 2 the first gene in a tandem array of two variable antigen genes is deleted, thus moving a previously silent gray gene next to a promoter. The recombination is between two short direct repeats common to both the black and gray genes. Although this results in the loss of that particular allele of the black gene as a nonreplicative circle, there usually would be another copy of the gray allele in the genome.
In the pathogens discussed here, the fourth general mechanism, multiple point mutations or conversion patches, usually occurs in a gene that has already been activated or moved by one of the other three mechanisms. In Figure 2, the expressed white gene undergoes limited gene conversion by the black gene, in a process similar to somatic mutations of rearranged immunoglobulin genes.
In any given strain, the repertoire may have considerable sequence diversity, but this does not mean that each strain has achieved a unique solution to the problem of immune avoidance. Other strains and other species in the same genus usually have a repertoire of genes homologous to the set of genes of the pathogen in question (11). The evolutionary distance between two variable antigen genes in the same pathogens may be greater than the distance between two genes in a different species. An example would be the hypothetical variable gene repertoire A, B, C, and D in species 1 and variable gene repertoire A', B', C', and D' in species 2. The sequence identity between A, B, C, and D in species may be no more than 40%, but there may be 80%-90% sequence identity between the B gene in species 1 and the B' gene in species 2.
The antigenic variation of relapsing fever spirochetes once attracted the attention of the early immunologists, such as Paul Ehrlich, because the infection proved the specificity of the immune response (8,9). In B. hermsii, a New World relapsing fever species, approximately 30 serotypes have been derived from a single cell (12). Specific antisera to these serotypes accounted for approximately 80%-90% of the variants that appeared during relapses of infection in mice in prospective experiments (13). The serotype of a Borrelia cell depends on its major surface antigen. The 30 or so antigens are divided approximately equally between two families: Variable Large Proteins (Vlp) of approximately 36 kDa and Variable Small Proteins (Vsp) of approximately 20 kDa (14-16). These abundant lipoproteins are anchored in the outer membrane by their lipid moieties. Although the vlp and vsp genes use the same locus for expression and may have identical signal peptides, no sequence homology can be identified between these two groups of proteins. Information about the variable antigens of other relapsing fever Borrelia spp. is less extensive, but B. recurrentis, the cause of louse-borne relapsing fever (17), B. crocidurae (18), an Old World relapsing fever species, and B. turicatae (19), another New World species, have vlp and vsp genes themselves. The Vsp proteins not only serve as variable antigens that may attract attention as an immune target but also determine tissue tropisms. In a clonal population of B. turicatae, expression of one vsp gene is associated with invasion of the central nervous system, while expression of another vsp gene is associated with high densities of spirochetes in the blood (20, 21).
B. hermsii uses all four general mechanisms for antigenic variation. Gene conversion between a linear plasmid containing a collection of silent vsp and vlp genes and another linear plasmid with an active vsp or vlp gene results in the replacement of one variable antigen gene with another downstream from a promoter (22,23). The gene conversion may be less extensive, yielding a chimeric vlp gene from a partial gene conversion (24). In B. turicatae, conversion may be more extensive, involving 10 or more kilobases downstream of the promoter (19). A DNA rearrangement in which the first member of a tandem pair on a single linear plasmid is deleted also yields a new serotype in the population (25). After the deletion, the formerly distal vsp in the pair is now next to the promoter. This type of rearrangement may be followed by a period of hypermutation at the 5' end of the gene (26). These frequent point mutations in the newly expressed gene further diversification of the vsp sequences.
These three types of events occur at a single expression site in the genome. Evidence also indicates that variation may also occur through the fourth mechanism: a change in transcription between two separate loci (27). A second locus for vsp expression has been found in B. hersmii on another linear plasmid (27). When this locus is active, the first expression site is silent (28). Activation of this second site is associated with infections of ticks, not mammals (29).
Borrelia burgdorferi and related species that cause Lyme disease have, as stated above, only one copy of the vsp orthologue, ospC, per cell, but it has several copies of sequences called vlsE genes, which are homologous to the Vlp proteins of relapsing fever Borrelia spp. (30). During infection, but not detectably in vitro, there is variation in expressed VlsE proteins through partial gene conversions from a tandem array of cassettes containing different hypervariable regions of vlsE sequences. Variants appeared in both immunodeficient as well as immunocompetent mice, but the rate of accumulation of amino acid changes in VlsE was higher in immunocompetent animals (31).
Anaplasmosis, a persistent intraerythrocytic infection of cattle and goats, has a global distribution. Infected animals have severe anemia and a higher rate of abortion. The infection, which is caused by members of the genus Anaplasma, an obligate intracellular rickettsia-like bacterium related to the genus Ehrlichia, is characterized by repetitive cycles of rickettsemia at 6- to 8-week intervals. In cattle infected with Anaplasma marginale, the number of pathogens in the blood varies between a peak of 106 to 107 per mL to a low of 102 per mL. In each cycle, the number of pathogens in the blood increases over 10 to 14 days and then precipitously declines (32). The cattle are reservoirs for the infection, and ixodid ticks are the vectors. Transmission to the vector ticks depends on the density of the pathogens in the blood (33).
Major surface protein 2 of A. marginale is an immunodominant outer membrane protein of approximately 40 kDa. Each strain has a large polymorphic family of msp2 genes; the variation occurs in the central region of the proteins. Multigene families of MSP2 paralogs have been found in Cowdria ruminatum (34), the cause of heartwater disease of ruminants in Africa and Caribbean, and Ehrlichia granulocytophila, the agent of human granulocytic ehrlichiosis (35). Each sequential cycle of rickettsemia is associated with a different transcript from at least 17 different msp2 genes in the family (36). Vaccinating animals with recombinant MSP2 produces antibodies specific for that MSP2. The genetic mechanisms for switches in msp2 genes are not known but may involve partial gene conversions through homologous recombination.
Trypanosoma brucei is a flagellated protozoon transmitted by tsetse flies to mammalian hosts, including humans and livestock. The infection consists of rising and falling parasitemia resulting from the generation of subpopulations that have antigenically different forms of a major variant-specific glycoprotein (VSG) at the cell surface (37). African trypanosomes switch at rates that are as low as 10-7 to 10-6 for syringe-passaged lines (38) or as high as 10-3 to 10-2 for field or fly-transmitted lines (39). VSG proteins, which are 400 to 500 amino acids in length, are anchored to the parasite's membrane at their carboxy terminus by a glycosyl-phospatidyl-inositol linkage. Besides immune evasion, other possible functions of VSGs include shielding other proteins (e.g., permeases) on the surface from immune attack and inhibiting phagocytosis (37).
A parasite can express several VSGs during infection in the mammalian host. Active genes for VSGs are located in one of 20 possible telomeric expression sites on the chromosomes and are transcribed with at least eight other genes (40,41), one of which encodes one of several variable transferrin receptors that confer different binding affinities for the transferrins of different mammals. Therefore, African trypanosomes combine antigenic variation of their surface coats with the ability to take up transferrin from their mammalian hosts (42).
A given VSG coat protein is encoded by a single vsg gene. Antigenic variation of VSG coats can occur by all the mechanisms described above, namely, transcriptional control, gene conversions, single cross-over events between telomeric genes, and point mutations (37). A complete VSG gene conversion is usual in the early stages of infection, while partial replacement and point mutations that may generate further diversity are observed in the more chronic stages (43,44). Short blocks of sequence homology in the upstream and downstream regions of the donor and acceptor genes may be required for the recombination events, but the precise basis for these switching events remains unknown.
A possible mechanism may involve the unusual DNA base J, which is enriched in silent telomeric sites but is absent in expressed regions (45). Site-specific nucleases have not been described, but RAD51, an enzyme involved in DNA break repair and genetic exchange in other eukaryotes, may be involved (46). For transcriptional activation and silencing of vsg expression in the bloodstream forms of African trypanosomes, the presence of a certain sequence within the promoter may not be critical. When an expression site vsg promoter is replaced by ribosomal DNA promoter, vsg expression sites may still be silenced or activated (47).
Plasmodium falciparum
During malaria infection, the apicomplexan parasites of the genus Plasmodium undergo repeated cycles of growth in erythrocytes. The species P. falciparum has strains that differ in several polymorphic proteins, but antigenic variation within a strain also occurs. The best-documented example of true antigenic variation is in the P. falciparum-infected erythrocyte membrane protein 1 (PfEMP1) antigens, which are expressed on the surface of the infected erythrocytes. Switching rates between PfEMP1 proteins may be as high as 10-2 per generation (Table) (48). By changing which PfEMP1 is expressed, the parasite evades the immune response directed against these immunodominant antigens. The PfEMP1 proteins also inhibit antigen presentation by dendritic cells and provide the means for the infected red cells to adhere to endothelium and extracellular matrix, thus avoiding clearance of the infected erythrocytes by the spleen (48-50).
The variable PfEMP1 proteins range from 200 to 350 kD. Their extracellular region has variable adhesive domains that confer the parasite-infected erythrocytes with a particular binding specificity that can include the extracellular matrix protein thrombospondin and a variety of endothelial receptors such as CD36, vascular cell adhesion molecule-1 (VCAM-1), E-selectin (ELAM-1), and intercellular cell adhesion molecule type 1 (ICAM-1) (49,50). These adhesive phenotypes lead to the sequestration of infected erythrocytes in the brain, lungs, kidneys, liver, or other organs, thereby determining the clinical manifestations of malaria.
The PfEMP1 proteins are encoded by members of the var family of genes (49,51,52). Each parasite devotes approximately 2% to 6% of its genomic DNA to a repertoire of 50 to 150 var genes clustered near the ends of chromosomes. Transcription of the var genes can occur from expression sites internal on the chromosomes or near a chromosome telomere (53). Changes in var expression appear to occur in situ by recombination-independent mechanisms (51,52). Evidence indicates that a single P. falciparum simultaneously transcribes multiple var genes during its early ring stages, but in trophozoites, tighter transcriptional control results in the expression of a single PfEMP-1 on the surface of the host cell (54,55).
Two additional variant multigene families that, like PfEMP1, are expressed on the surface of infected red blood cells, induce specific antibodies, and undergo clonal variation have been described recently (56,57). These proteins are encoded by the rif and STEVOR genes, which are located near the telomeres that contain the var genes.
Babesia bovis
Members of the genus Babesia cause one of the most common parasitic infections worldwide in wild and domestic animals. Some of the species, such as B. microti, have been transmitted to humans. Like Plasmodium, Babesia are intraerythrocytic parasites, but they are transmitted by ticks, not mosquitoes. While several multigene families have been described for various species of Babesia, clonal antigenic variation of B. bovis, a parasite of cattle, is best documented (58). The variant erythrocyte surface antigen (VESA1) of B. bovis is a heterodimeric protein expressed on the surface of infected red blood cells. The rapid variation of these polymorphic proteins likely contributes to chronic infection in cattle by prolonging the parasite's survival through immune evasion and sequestration of the infected red blood cells in peripheral organs (58). The VESA1 proteins, which have an approximate molecular weight of 128 kDa (59), are expressed on the external tips of the membrane knobs of infected erythrocytes. Their cytoadhesive phenotype depends on the antigenic and structural changes of the VESA1 proteins (60). The gene encoding the VESA1a subunit has been recently shown to belong to the ves multigene family (61). The predicted protein does not seem to have cleavable signal sequence, but it does have a predicted transmembrane segment and a cysteine/lysine-rich domain (61). The molecular events that determine the switching mechanism in B. babesia are unknown.
The requirement for vector transmission of these infectious pathogens provides a powerful selection for mechanisms that prolong parasitemia. Through convergent evolution, several vector-borne pathogens have arrived at the same strategy of antigenic variation to achieve this goal. The similarity in the genetic mechanisms that such unrelated pathogens as African trypanosomes and relapsing fever Borrelia spp. use for antigenic variation is remarkable.
Antigenic variation has important implications for the development of vaccines against these pathogens. If the variable antigen is to be the target of immunoprophylaxis, the vaccine would likely need to be multivalent, perhaps to the point of impracticality. If the infected host animal has not solved the problem of identifying an antigen that is conserved among the variants, thereby neutralizing the infection earlier, how can vaccine developers hope to do this?
A possible way to meet this challenge is to focus on the function domains of the variable proteins. The variable antigens of both the bacterial and parasite pathogens have other roles in pathogenesis besides immune evasion. These include tissue tropism, shielding of adjacent molecules, inhibition of phagocytosis, modulation of antigen presentation, and selective adherence. Certain regions of the variable protein may be irrelevant for these functions of the pathogen, and consequently the encoding DNA sequences could be highly divergent among alleles. On the other hand, the regions conferring these functions would likely be more constrained in structure and thus comparatively more susceptible to cross-reacting antibodies.
Another possible way to meet the challenge of antigenic variation is to focus on the vector-specific surface antigens of these pathogens. The repertoire expressed in the arthropod vector, which lacks an adaptive immune system, is generally more limited than that expressed in the vertebrate host. The Lyme disease vaccine is an example of successful targeting of a vector-specific protein. Although B. burgdorferi has not yet been proven to undergo true antigenic variation, there is considerable diversity in the ospC sequences that define strain identity within a given area in which transmission to humans occurs. A vaccine based on OspC would likely need to be multivalent. In contrast, B. burgdorferi's OspA protein (62), the sole protein in the vaccine, is natively expressed in the tick's midgut but usually not during infection of mammals (63). Perhaps because of OspA's infrequent encounters with the mammalian adaptive immune system in nature, there is little divergence in ospA sequences between strains of B. burgdorferi (64). The OspA-based vaccine apparently works by eliciting antibodies that kill or inhibit the spirochetes in the tick, before expression of the more polymorphic ospC and vlsE genes in the mammalian host (65).
Dr. Barbour is professor of medicine and microbiology & molecular genetics at the University of California Irvine. His research focuses on the molecular pathogenesis of relapsing fever and Lyme disease and the prevention of tick-borne diseases.
Dr. Restrepo is head of the molecular parasitology group at the Corporación para Investigaciones Biológicas in Medellín, Colombia. Her current research interests focus on the molecular and immunologic aspects of neurocysticercosis.
Acknowledgment
This research was supported by grants AI24424 and AI37248 from the National Institutes of Health and a grant from the Centers for Disease Control and Prevention (A.G.B.) and by grants from the Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología Francisco José de Caldas, Fogarty International, and the International Society for Infectious Diseases (B.I.R.).
References
- Wang IN, Dykhuizen DE, Qiu W, Dunn JJ, Bosler EM, Luft BJ. Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics. 1999;151:15–30.PubMedGoogle Scholar
- Stevenson B, Bockenstedt LK, Barthold SW. Expression and gene sequence of outer surface protein C of Borrelia burgdorferi reisolated from chronically infected mice. Infect Immun. 1994;62:3568–71.PubMedGoogle Scholar
- Beale GH. J.F. W. Antigenic variation in unicellular organisms. Annu Rev Microbiol. 1961;15:263–96. DOIGoogle Scholar
- Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev. 1986;50:381–400.PubMedGoogle Scholar
- de la Vega I, Gonzalez A, Blasco R, Calvo V, Vinuela E. Nucleotide sequence and variability of the inverted terminal repetitions of African swine fever virus DNA. Virology. 1994;201:152–6. DOIPubMedGoogle Scholar
- Zuckert WR, Meyer J, Barbour AG. Comparative analysis and immunological characterization of the Borrelia Bdr protein family. Infect Immun. 1999;67:3257–66.PubMedGoogle Scholar
- Turner CM. Antigenic variation in Trypanosoma brucei infections: an holistic view. J Cell Sci. 1999;112:3187–92.PubMedGoogle Scholar
- Barbour AG. Immunobiology of relapsing fever. Contrib Microbiol Immunol. 1987;8:125–37.PubMedGoogle Scholar
- Vickerman K. Trypanosome sociology and antigenic variation. Parasitology. 1989;99:S37–47. DOIPubMedGoogle Scholar
- Deitsch KW, Moxon ER, Wellems TE. Shared themes of antigenic variation and virulence in bacterial, protozoal, and fungal infections. Microbiol Mol Biol Rev. 1997;61:281–93.PubMedGoogle Scholar
- Hinnebusch BJ, Barbour AG, Restrepo BI, Schwan TG. Population structure of the relapsing fever spirochete Borrelia hermsii as indicated by polymorphism of two multigene families that encode immunogenic outer surface lipoproteins. Infect Immun. 1998;66:432–40.PubMedGoogle Scholar
- Barbour AG. Linear DNA of Borrelia species and antigenic variation. Trends Microbiol. 1993;1:236–9. DOIPubMedGoogle Scholar
- Stoenner HG, Dodd T, Larsen C. Antigenic variation of Borrelia hermsii. J Exp Med. 1982;156:1297–311. DOIPubMedGoogle Scholar
- Barbour AG, Tessier SL, Stoenner HG. Variable major proteins of Borrellia hermsii. J Exp Med. 1982;156:1312–24. DOIPubMedGoogle Scholar
- Burman N, Bergstrom S, Restrepo BI, Barbour AG. The variable antigens Vmp7 and Vmp21 of the relapsing fever bacterium Borrelia hermsii are structurally analogous to the VSG proteins of the African trypanosome. Mol Microbiol. 1990;4:1715–26. DOIPubMedGoogle Scholar
- Restrepo BI, Kitten T, Carter CJ, Infante D, Barbour AG. Subtelomeric expression regions of Borrelia hermsii linear plasmids are highly polymorphic. Mol Microbiol. 1992;6:3299–311. DOIPubMedGoogle Scholar
- Vidal V, Scragg IG, Cutler SJ, Rockett KA, Fekade D, Warrell DA, Variable major lipoprotein is a principal TNF-inducing factor of louse-borne relapsing fever. Nat Med. 1998;4:1416–20. DOIPubMedGoogle Scholar
- Shamaei-Tousi A, Martin P, Bergh A, Burman N, Brännström T, Bergström S. Erythrocyte-aggregating relapsing fever spirochete Borrelia crocidurae induces formation of microemboli. J Infect Dis. 1999;180:1929–38. DOIPubMedGoogle Scholar
- Pennington PM, Cadavid D, Bunikis J, Norris SJ, Barbour AG. Extensive interplasmidic duplications change the virulence phenotype of the relapsing fever agent Borrelia turicatae. Mol Microbiol. 1999;34:1120–32. DOIPubMedGoogle Scholar
- Cadavid D, Thomas DD, Crawley R, Barbour AG. Variability of a bacterial surface protein and disease expression in a possible mouse model of systemic Lyme borreliosis. J Exp Med. 1994;179:631–42. DOIPubMedGoogle Scholar
- Pennington PM, Allred CD, West CS, Alvarez R, Barbour AG. Arthritis severity and spirochete burden are determined by serotype in the Borrelia turicatae-mouse model of Lyme disease. Infect Immun. 1997;65:285–92.PubMedGoogle Scholar
- Kitten T, Barbour AG. Juxtaposition of expressed variable antigen genes with a conserved telomere in the bacterium Borrelia hermsii. Proc Natl Acad Sci U S A. 1990;87:6077–81. DOIPubMedGoogle Scholar
- Barbour AG, Burman N, Carter CJ, Kitten T, Bergstrom S. Variable antigen genes of the relapsing fever agent Borrelia hermsii are activated by promoter addition. Mol Microbiol. 1991;5:489–93. DOIPubMedGoogle Scholar
- Kitten T, Barrera AV, Barbour AG. Intragenic recombination and a chimeric outer membrane protein in the relapsing fever agent Borrelia hermsii. J Bacteriol. 1993;175:2516–22.PubMedGoogle Scholar
- Restrepo BI, Carter CJ, Barbour AG. Activation of a vmp pseudogene in Borrelia hermsii: an alternate mechanism of antigenic variation during relapsing fever. Mol Microbiol. 1994;13:287–99. DOIPubMedGoogle Scholar
- Restrepo BI, Barbour AG. Antigen diversity in the bacterium B. hermsii through "somatic" mutations in rearranged vmp genes. Cell. 1994;78:867–76. DOIPubMedGoogle Scholar
- Carter CJ, Bergstrom S, Norris SJ, Barbour AG. A family of surface-exposed proteins of 20 kilodaltons in the genus Borrelia. Infect Immun. 1994;62:2792–9.PubMedGoogle Scholar
- Barbour AG, Carter CJ, Sohaskey CD. Surface protein variation by expression site switching in the relapsing fever agent Borrelia hermsii. Infect Immun. 2000. In press.
- Schwan TG, Hinnebusch BJ. Bloodstream- versus tick-associated variants of a relapsing fever bacterium. Science. 1998;280:1938–40. DOIPubMedGoogle Scholar
- Zhang JR, Hardham JM, Barbour AG, Norris SJ. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997;89:275–85. DOIPubMedGoogle Scholar
- Zhang JR, Norris SJ. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun. 1998;66:3689–97.PubMedGoogle Scholar
- Kieser ST, Eriks IS, Palmer GH. Cyclic rickettsemia during persistent Anaplasma marginale infection of cattle. Infect Immun. 1990;58:1117–9.PubMedGoogle Scholar
- Palmer GH, Abbott JR, French DM, McElwain TF. Persistence of Anaplasma ovis infection and conservation of the msp-2 and msp-3 multigene families within the genus Anaplasma. Infect Immun. 1998;66:6035–9.PubMedGoogle Scholar
- Sulsona CR, Mahan SM, Barbet AF. The map1 gene of Cowdria ruminantium is a member of a multigene family containing both conserved and variable genes. Biochem Biophys Res Commun. 1999;257:300–5. DOIPubMedGoogle Scholar
- Zhi N, Ohashi N, Rikihisa Y. Multiple p44 genes encoding major outer membrane proteins are expressed in the human granulocytic ehrlichiosis agent. J Biol Chem. 1999;274:17828–36. DOIPubMedGoogle Scholar
- French DM, Brown WC, Palmer GH. Emergence of Anaplasma marginale antigenic variants during persistent rickettsemia. Infect Immun. 1999;67:5834–40.PubMedGoogle Scholar
- Rudenko G, Cross M, Borst P. Changing the end: antigenic variation orchestrated at the telomeres of African trypanosomes. Trends Microbiol. 1998;6:113–6. DOIPubMedGoogle Scholar
- Lamont GS, Tucker RS, Cross GA. Analysis of antigen switching rates in Trypanosoma brucei. Parasitology. 1986;92:355–67. DOIPubMedGoogle Scholar
- Turner CM. The rate of antigenic variation in fly-transmitted and syringe-passaged infections of Trypanosoma brucei. FEMS Microbiol Lett. 1997;153:227–31. DOIPubMedGoogle Scholar
- Pays E, Tebabi P, Pays A, Coquelet H, Revelard P, Salmon D, The genes and transcripts of an antigen gene expression site from T. brucei. Cell. 1989;57:835–45. DOIPubMedGoogle Scholar
- Lips S, Revelard P, Pays E. Identification of a new expression site-associated gene in the complete 30.5 kb sequence from the AnTat 1.3A variant surface protein gene expression site of Trypanosoma brucei. Mol Biochem Parasitol. 1993;62:135–7. DOIPubMedGoogle Scholar
- Bitter W, Gerrits H, Kieft R, Borst P. The role of transferrin-receptor variation in the host range of Trypanosoma brucei. Nature. 1998;391:499–502. DOIPubMedGoogle Scholar
- Baltz T, Giroud C, Bringaud F, Eisen H, Jacquemot C, Roth CW. Exposed epitopes on a Trypanosoma equiperdum variant surface glycoprotein altered by point mutations. EMBO J. 1991;10:1653–9.PubMedGoogle Scholar
- Lu Y, Alarcon CM, Hall T, Reddy LV, Donelson JE. A strand bias occurs in point mutations associated with variant surface glycoprotein gene conversion in Trypanosoma rhodesiense. Mol Cell Biol. 1994;14:3971–80.PubMedGoogle Scholar
- Gommers-Ampt J, Lutgerink J, Borst P. A novel DNA nucleotide in Trypanosoma brucei only present in the mammalian phase of the life-cycle. Nucleic Acids Res. 1991;19:1745–51. DOIPubMedGoogle Scholar
- McCulloch R, Barry JD. A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 1999;13:2875–88. DOIPubMedGoogle Scholar
- Rudenko G, Blundell PA, Dirks-Mulder A, Kieft R, Borst P. A ribosomal DNA promoter replacing the promoter of a telomeric VSG gene expression site can be efficiently switched on and off in T. brucei. Cell. 1995;83:547–53. DOIPubMedGoogle Scholar
- Roberts DJ, Craig AG, Berendt AR, Pinches R, Nash G, Marsh K, Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature. 1992;357:689–92. DOIPubMedGoogle Scholar
- Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. DOIPubMedGoogle Scholar
- Gardner JP, Pinches RA, Roberts DJ, Newbold CI. Variant antigens and endothelial receptor adhesion in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1996;93:3503–8. DOIPubMedGoogle Scholar
- Smith JD, Chitnis CE, Craig AG, Roberts DJ, Hudson-Taylor DE, Peterson DS, Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. 1995;82:101–10. DOIPubMedGoogle Scholar
- Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100. DOIPubMedGoogle Scholar
- Hernandez-Rivas R, Mattei D, Sterkers Y, Peterson DS, Wellems TE, Scherf A. Expressed var genes are found in Plasmodium falciparum subtelomeric regions. Mol Cell Biol. 1997;17:604–11.PubMedGoogle Scholar
- Chen Q, Fernandez V, Sundstrom A, Schlichtherle M, Datta S, Hagblom P, Developmental selection of var gene expression in Plasmodium falciparum. Nature. 1998;394:392–5. DOIPubMedGoogle Scholar
- Scherf A, Hernandez-Rivas R, Buffet P, Bottius E, Benatar C, Pouvelle B, Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum. EMBO J. 1998;17:5418–26. DOIPubMedGoogle Scholar
- Kyes SA, Rowe JA, Kriek N, Newbold CI. Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc Natl Acad Sci U S A. 1999;96:9333–8. DOIPubMedGoogle Scholar
- Fernandez V, Hommel M, Chen Q, Hagblom P, Wahlgren M. Small, clonally variant antigens expressed on the surface of the Plasmodium falciparum-infected erythrocyte are encoded by the rif gene family and are the target of human immune responses. J Exp Med. 1999;190:1393–404. DOIPubMedGoogle Scholar
- Allred DR, Cinque RM, Lane TJ, Ahrens KP. Antigenic variation of parasite-derived antigens on the surface of Babesia bovis-infected erythrocytes. Infect Immun. 1994;62:91–8.PubMedGoogle Scholar
- O'Connor RM, Lane TJ, Stroup SE, Allred DR. Characterization of a variant erythrocyte surface antigen (VESA1) expressed by Babesia bovis during antigenic variation. Mol Biochem Parasitol. 1997;89:259–70. DOIPubMedGoogle Scholar
- O'Connor RM, Allred DR. Selection of Babesia bovis-infected erythrocytes for adhesion to endothelial cells coselects for altered variant erythrocyte surface antigen isoforms. J Immunol. 2000;164:2037–45.PubMedGoogle Scholar
- Allred DR, Carlton JM, Satcher RL, Long JA, Brown WC, Patterson PE, The ves multigene family of B. bovis encodes components of rapid antigenic variation at the infected erythrocyte surface. Mol Cell. 2000;5:153–62. DOIPubMedGoogle Scholar
- Barbour AG, Tessier SL, Todd WJ. Lyme disease spirochetes and ixodid tick spirochetes share a common surface antigenic determinant defined by a monoclonal antibody. Infect Immun. 1983;41:795–804.PubMedGoogle Scholar
- Schwan TG, Piesman J. Temporal changes in outer surface proteins A and C of the Lyme disease-associated spirochete, Borrelia burgdorferi, during the chain of infection in ticks and mice. J Clin Microbiol. 2000;38:382–8.PubMedGoogle Scholar
- Jonsson M, Noppa L, Barbour AG, Bergstrom S. Heterogeneity of outer membrane proteins in Borrelia burgdorferi: comparison of osp operons of three isolates of different geographic origins. Infect Immun. 1992;60:1845–53.PubMedGoogle Scholar
- de Silva AM, Zeidner NS, Zhang Y, Dolan MC, Piesman J, Fikrig E. Influence of outer surface protein A antibody on Borrelia burgdorferi within feeding ticks. Infect Immun. 1999;67:30–5.PubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 6, Number 5—October 2000
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Alan Barbour, Department of Microbiology & Molecular Genetics, University of California Irvine, Irvine, CA 92697-4025; fax 949-824-5626
Top