Volume 19, Number 10—October 2013
Research
Genetic Recombination and Cryptosporidium hominis Virulent Subtype IbA10G2
Cite This Article
Citation for Media
Abstract
Little is known about the emergence and spread of virulent subtypes of Cryptosporidium hominis, the predominant species responsible for human cryptosporidiosis. We conducted sequence analyses of 32 genetic loci of 53 C. hominis specimens isolated from a longitudinally followed cohort of children living in a small community. We identified by linkage disequilibrium and recombination analyses only limited genetic recombination, which occurred exclusively within the 60-kDa glycoprotein gene subtype IbA10G2, a predominant subtype for outbreaks in industrialized nations and a virulent subtype in the study community. Intensive transmission of virulent subtype IbA10G2 in the study area might have resulted in genetic recombination with other subtypes. Moreover, we identified selection for IbA10G2 at a 129-kb region around the 60-kDa glycoprotein gene in chromosome 6. These findings improve our understanding of the origin and evolution of C. hominis subtypes and the spread of virulent subtypes.
Cryptosporidium spp. are emerging pathogens of humans and a variety of vertebrates, and cause severe diarrhea in immunocompetent and immunocompromised persons (1). Cryptosporidium hominis is responsible for >70% of human infections in most areas, especially North America and developing countries (2). C. hominis is primarily transmitted anthroponotically, has several transmission routes, and causes numerous waterborne outbreaks of diarrheal illness each year in the United States and other industrialized nations.
Among several C. hominis subtype groups (e.g., Ia, Ib, Id, Ie, If, Ig) identified by sequence analysis of the 60-kDa glycoprotein (gp60) gene, the Ib subtype is the major subtype responsible for waterborne and foodborne outbreaks of cryptosporidiosis in many countries. Subtype IbA10G2 has been found in ≈50% of C. hominis–associated outbreaks in the United States, including the massive outbreak in Milwaukee, Wisconsin, USA, in 1993 (2,3). It is the only subtype identified in cryptosporidiosis outbreaks by C. hominis in countries in Europe and in Australia (4–11). In a longitudinal birth-cohort study of cryptosporidiosis in a periubran shantytown in Lima, Peru, IbA10G2 was more virulent than other C. hominis subtypes (12). Genetic determinants for virulence of Cryptosporidium spp. and reasons for emergence of virulent subtypes are poorly understood because of availability of only limited genomic sequence data and lack of robust cultivation systems and genetic manipulation tools (13).
We conducted a comparative population genetic analysis of virulent C. hominis subtype IbA10G2 in children living in a periurban community in Lima, Peru, by multilocus sequence typing (MLST) of 32 genetic markers. Data obtained should be useful in understanding emergence and spread of virulent C. hominis subtypes.
Specimens, Species, and Subtype Determination
Fecal specimens microscopically positive for Cryptosporidium spp. were collected during a longitudinal cohort study of enteric diseases in children living in a periurban shantytown in Lima, Peru, during 2004–2006. The field study was conducted in the same community used in a previous longitudinal cohort study of enteric pathogens and had a similar study design (12), except that children enrolled in this study were older (mean age 3.43 years vs. 14 days). The study was approved by the institutional review boards of Johns Hopkins University and the Centers for Diseases Control and Prevention.
DNA was extracted from 200 μL of microscopy-positive specimens by using the FastDNA SPIN Kit for Soil (MP Biomedicals, Irvine, CA, USA). Cryptosporidium spp. in specimens were genetically characterized at the species level by PCR–restriction fragment length polymorphism analysis of the small subunit rRNA gene (14) and at the subtype level by DNA sequence analysis of the gp60 gene (15). A total of 53 C. hominis–positive specimens (1 specimen/child) belonging to 4 gp60 subtype groups were selected for MLST analysis: Ia (9 specimens of IaA13R8 and 1 specimen of IaA13R7), Ib (26 specimens of IbA10G2), Id (6 specimens of IdA10 and 5 specimens of IdA20), and Ie (6 specimens of IeA11G3T3). No specimens used in the study had mixed C. hominis subtypes.
MLST Markers
Among the 8 chromosomes of Cryptosporidium spp., chromosome 6 was fully sequenced for 2 C. parvum specimens and 1 C. hominis specimen, which facilitated search for additional microsatellite and minisatellite markers for population genetics analysis. The 8 polymorphic markers on chromosome 6 (gp60, CP47, CP56, MSC6–5, MSC6–7, TSP8, Mucin1, and DZ-HRGP) used in several MLST and population genetic analyses of C. hominis and C. parvum (16–18) were included in this study. We searched for additional microsatellite and minisatellite markers in the chromosome 6 genome by using Tandem Repeats Finder software (http://tandem.bu.edu/trf/trf.html). Of 325 short tandem repeat sequences identified, 46 loci were initially selected at spaced intervals that covered the entire chromosome 6. A nested PCR approach was used for amplification of all potential targets. After an initial evaluation of primers by PCR and DNA sequencing of IaA15R3, IaA20R3, IbA10G2, IdA10, and IeA11G3T3 subtypes of C. hominis from the United States and Peru, 24 polymorphic loci (Table 1, Appendix), and the 8 markers previously identified, were used in this study.
MLST PCR and Sequencing
Conditions for nested PCR analysis of each marker were similar to those described (16–18), except for differences in annealing temperatures specified in Table 1. One negative control and 1 positive control (C. parvum IIaA16G2R1 subtype) were used in each PCR. PCR products of the expected size were sequenced in both directions by using an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences obtained from each locus were edited and aligned by using BioEdit 7.04 version (www.mbio.ncsu.edu/BioEdit/bioedit.html) and ClustalX version 1.81 (www.clustal.org/), respectively. Nucleotide sequences of all haplotypes of polymorphic loci were deposited in GenBank under accession nos. JX088398–JX088417 and JX088427–JX088501.
Analyses of Sequence Polymorphism
For each marker, sequences of 53 C. hominis specimens were used for calculation of haplotype diversity (Hd), number of haplotypes, number of polymorphic and segregating sites, intragenic linkage disequilibrium (LD), and intragenic recombination rates by using DnaSP version 5.10.00 (www.ub.es/dnasp/). The ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site was calculated for gp60 and several nearby loci (C6–830′, C6–1000′, C6–1420, CP47, and C6–2600) by using DnaSP. Thereafter, sequences of polymorphic loci were concatenated as a single multilocus contig to calculate gene diversity, interlocus LD, and recombination rates by using DnaSP. Pairwise intergenic LD was also evaluated among polymorphic loci by using the Fisher exact test and Markov chain parameters in Arlequin version 3.1 (http://cmpg.unibe.ch/software/arlequin3/).
Substructure Analysis
Population substructures were analyzed by using the Bayesian analysis tool STRUCTURE version 2.2 (http://pritch.bsd.uchicago.edu/structure.html). Conversion of microsatellite and minisatellite sequences to allelic data was performed by using the Excel Microsatellite Tool Kit 3.1.1 (http://animalgenomics.ucd.ie/sdepark/ms-toolkit/) and Genepop version 4.0 (http://genepop.curtin.edu.au/). Several analyses of allelic data were performed by using K (likely populations) ranging from 2 to 8 and 50,000 iterations after a burn-in of 50,000 iterations. Output at K = 2–5 provided the best fit to MLST data and were used in further analyses. To provide an alternative view of substructuring, a median-joining algorithm analysis (19) was conducted by using Network version 4.6.1.0 (www.fluxus-ngineering.com/sharenet.htm). Each gp60 subtype was further analyzed for LD and recombination by using DnaSP and Arlequin. Population differentiation (FST) between IbA10G2 and non-IbA10G2 subtypes was also assessed at each genetic locus by using Arlequin.
Multilocus Gene Diversity
Sequence polymorphism among the 53 C. hominis specimens was detected at 25 loci (Table 2), including variations in copy numbers of microsatellite or minisatellite repeat and single nucleotide substitutions (SNPs) and insertion and deletion (indels) in the nonrepeat regions. The remaining 7 loci (C6–190, C6–500′, TSP8, C6–830, C6–870, MSC6–5, and Mucin1) were monomorphic in the study population. The Hd of individual polymorphic locus ranged from 0.0377 to 0.9231, and most loci with higher Hd were situated in the first 25% of chromosome 6 (Table 2). Number of haplotypes per locus ranged from 2 to 21, and more haplotypes were at loci C6–60 (21), CP47 (6), gp60 (6), C6–230 (6), and C6–740 (5). MLST analysis confirmed the absence of concurrence of mixed C. hominis subtypes in the 53 specimens.
Alignment of combined multilocus sequences covered 15,717 bp and 968 polymorphic sites, including 362 segregating sites and 606 indels. Multilocus sequences had 43 multilocus genotypes (MLGs), an Hd of 0.9898, and a nucleotide diversity of 0.339. The frequency of MLGs ranged from 7.5% (1 MLG with 4 specimens), 5.7% (1 MLG with 3 specimens), and 3.8% (5 MLGs each with 2 specimens) to 1.9% (36 MLGs each with 1 specimen). Because 431/968 polymorphic sites and 297/362 segregating sites occurred within the gp60 locus, a second analysis was performed by using concatenated sequences excluding gp60 (14,768 bp) with 537 polymorphic sites, including 65 segregating sites and 472 indel sites. A lower nucleotide diversity of 0.289 was observed. However, the number of MLGs (43) and haplotype diversity (0.9898) remained the same.
LD and Recombination
Intragenic LD between pairs of segregating sites was assessed for each polymorphic locus. The analysis was possible only for 7 loci (C6–60, C6–280, C6–350, C6–1000′, gp60, CP47, and C6–4110) that had at ≥3 segregating sites. Incomplete intragenic LD (|D′| Y = 0.9729 + 0.0130X), where Y is the LD value and X is the nucleotide distance in kb, was observed at the gp60 locus, and complete intragenic LD (|D′| Y = 1.0000 + 0.0000X) was found at each of the 6 remaining loci (Table 2). An intragenic recombination test identified 26 potential recombination events (Rms) at the gp60 locus and no recombination at the remaining loci (Table 2).
On the basis of the concatenated multilocus sequence data, interlocus LD was assessed over all segregating sites by using pairwise comparisons (20). Analysis of combined multilocus sequences of all loci resulted in an overall interlocus genetic association (Zns) value of 0.2711 (95% CI 0.0695–0.3918; the probability p for expected Zns ≤0.2711 was 0.883). Of 47,895 pairwise comparisons, 25,904 were significant by Fisher exact test, and 8,714 were significant after Bonferroni correction (Table 3). In an additional LD analysis per site, strong but incomplete LD (|D′| Y = 0.9289 – 0.0299X) was detected with a negative slope, indicating a decrease in linkage with increased nucleotide distance (Table 3). When analysis was performed after excluding gp60, the test produced a Zns value of 0.2184 (95% CI 0.0654–0.4469, p = 0.759). Of 2,080 pairwise comparisons, 921 were significant by Fisher exact test, and 333 were significant after Bonferroni correction. Incomplete LD (|D′| Y = 0.7886 + 0.0051X) was also observed in LD analysis per site (Table 3). Thus, recombination might occur because of incomplete LD. An overall recombination test showed a minimum of 33 potential recombination events and an estimated 5.0 R/gene (Table 3). When analysis was performed after exclusion of gp60, only 6 recombination events and 1.8 R/gene were observed (Table 3). Thus, recombination was occurring mostly at the gp60 locus.
Substructure in C. hominis
Figure 1

Figure 1. . Relationship among various 60-kDa glycoprotein gene subtypes of Cryptosporidium hominis by substructure analysis. Predicted population numbers K = 2–5 were applied in STRUCTURE version 2.2 (http://pritch.bsd.uchicago.edu/structure.html) analysis of the data. Colored...
The evolutional relationship among gp60 subtypes of C. hominis was inferred by using STUCTURE and predicted population numbers K = 2–5. The ancestral population size K = 5 was considered the best estimate of current population substructure (Figure 1). The most dominant feature in the output was heterogeneity of specimens belonging to subtype IbA10G2 compared with other subtypes. Within 26 specimens of IbA10G2, the pattern of combinations in STRUCTURE suggested a mixture of ancestral types, reflecting likely genetic recombination in the subtype. In contrast, the 10 specimens of the Ia subtype (including 9 specimens of IaA13R8 and 1 specimen of IaA13R7) had mostly a single pattern, thus providing strong support for it being a separate population with a unique ancestry. Likewise, intrasubtype homogeneity was also observed in 6 specimens of IdA10, five specimens of IdA20, and 6 specimens of IeA11G3T3. Within the Id subtype, specimens of IdA10 and IdA20 showed different patterns regardless of the K value, indicating that these 2 Id subtypes had distinct ancestries with little mixture and genetic recombination. On the basis of patterns at K = 5 (Figure 1), some IbA10G2 specimens had a mixture of patterns, which were the dominant patterns in subtype IaA13R8 and subtype IeA11G3T3, respectively, suggesting that these IbA10G2 specimens might have resulted from genetic exchange between the ancestor of subtype IbA10G2 and the ancestor of subtype IaA13R8 or IeA11G3T3.
Figure 2
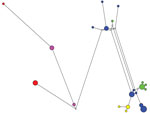
Figure 2. . Median-joining network for various subtypes of Cryptosporidium hominis. Circles are proportional to the frequency of each multilocus genotype (MLG) (21 MLGs on the basis of segregating sites of concatenated sequences...
We also conducted a median-joining network analysis of the MLST data for the 53 specimens. Among 21 MLGs generated on the basis of segregating sites of concatenated sequences, excluding gp60, we did not find any shared MLGs between gp60 subtypes of C. hominis (Figure 2). Compared with STRUCTURE analyses, the results of network analysis showed similarity to patterns at K = 2 (Figure 1), and supported the conclusion on the heterogeneity of subtype IbA10G2. In network analysis, central types are usually possible ancestors, and peripheral types are descendants (21). Because we did not find any MLG as the central type, it was impossible to define a single ancestral line that gave rise to other lines. This finding could be caused by the small sample size and few MLGs in this study.
Comparative Population Genetics of IbA10G2
Figure 3

The population genetics of various gp60 subtypes was assessed by analyses of LD and recombination rates. In pairwise interlocus LD analysis of multilocus sequences including or excluding gp60, strong but incomplete LD (|D′| Y = 0.9342 + 0.0001X or |D′| Y = 0.9302 + 0.0010X) was observed in subtype IbA10G2, suggesting recombination within this subtype. In contrast, all other subtypes showed complete LD (|D′| Y = 1.0000 + 0.0000X) among all sites, indicative of no genetic recombination within these subtypes (Table 3). In addition, pairwise intergenic LD was also evaluated between 25 loci, resulting in 87 instances of significant pairwise LD (p<0.05) observed in subtype IbA10G2 compared with 227 significant LD in the remaining subtypes (Figure 3). In IbA10G2 specimens, loci around gp60 (locus 12) had no LD with any other loci in chromosome 6. This finding indicated genetic recombination in subtype IbA10G2 at loci near gp60.
To validate these observations, recombination tests were conducted for all gp60 subtypes. Using full multilocus sequences, we found that among 33 potential Rms observed in overall recombination analysis, 4 Rms were detected in subtype IbA10G2 but no Rm was found within each of the remaining subtypes (Table 3). When analysis was performed that excluded gp60, among 6 potential Rms in the overall recombination analysis, 4 Rms were still detected in subtype IbA10G2 but no Rm was seen in the other subtypes (Table 3). Thus, genetic recombination occurred only within the subtype IbA10G2, a conclusion in agreement with results of the STRUCTURE and LD analyses.
Figure 4

The genetic determinant for differences between IbA10G2 and non-IbA10G2 subtypes was assessed by comparison of haplotype diversity of the 2 groups at all 25 polymorphic loci in chromosome 6. At most of the genetic loci, the haplotype diversity of the 2 groups was similar, and there was no clear population differentiation at these loci (Figure 4, Table 4). However, at 4 loci (C6–830′, C6–1000′, gp60, C6–1420) around gp60, an absence of genetic diversity was observed in subtype IbA10G2 compared with non-IbA10G2 subtypes (Figure 4). The region (129 kb) of homogeneity in subtype IbA10G2 was located in a 210–339-kb region in chromosome 6. Pairwise FST analysis showed highly significant differentiation between the IbA10G2 subtype and non-IbA10G2 subtypes at 4 loci around gp60, C6–1000′ (p<0.00001), gp60 (p<0.00001), CP47 (p<0.00001), and C6–2600 (p<0.001). The only other locus that showed such a level of differentiation between the 2 groups was C6–3520 (Table 4).
Like other apicomplexan parasites, the life cycle of Cryptosporidium spp. has a sexual phase, during which sexual recombination can occur between genetically distinct strains (22). C. parvum, the species that infects humans and some animals, undergoes meiotic recombination between different lineages in genetic-crossing experiments (23,24). LD analyses of natural C. parvum populations have also shown genetic recombination in most study areas (22,25–28). In contrast, C. hominis is primarily infectious to humans, and previous MLST studies showed a clonal population structure. Genetic recombination was believed to be rare or nonexistent in C. hominis (17,25,28–30). However, the small number of markers used in previous studies might have resulted in relatively low resolution in population structure analysis, which could have led to failure in detecting genetic recombination in C. hominis. In the present study, we examined population substructure of C. hominis in a cohort of children living in a small study area by using 32 genetic markers.
On the basis of multilocus sequence data and allelic profiles for 53 specimens, analysis showed strong LD among 25 polymorphic loci, suggesting an overall nonpanmictic population structure of C. hominis. A recombination test showed only limited genetic recombination at the gp60 locus. Thus, the high level of LD and limited recombination found in the overall population could be explained by an essential clonal population structure of C. hominis in the field site in Peru, which is consistent with information on C. hominis population genetics in other countries (17,25,28,29).
Results of LD and recombination analyses suggest that limited recombination in the study population of C. hominis occurred mostly at gp60 or loci around gp60. Thus far, the gp60 gene is the most polymorphic marker identified in the Cryptosporidium genome. Because of its high sequence heterogeneity, gp60 has become the most widely used gene in Cryptosporidium spp. subtyping, which categorizes C. hominis and C. parvum into several subtype groups and various subtypes within each subtype group (2). The gp60 gene encodes glycoproteins gp15 and gp45, which are implicated in attachment to and invasion of host cells (31,32). Because attachment of sporozoites to epithelial cells and invasion of the host cell membrane are crucial steps in the pathogenesis of cryptosporidiosis, these 2 glycoproteins are presumed to be surface-associated virulence determinants that may be under host immune selection, which might explain the extensive polymorphism in the gp60 gene (27). Genetic recombination appears to be associated with high sequence polymorphism in the gp60 gene (27). However, a less speculative understanding of the role the gp60 gene in pathogenesis of cryptosporidiosis requires further investigations.
Recent studies have suggested that telomeric/subtelomeric regions are highly polymorphic and might encode putative virulence factors (33,34). However, these studies did not compare phenotypic differences among isolates, and data for the present study do not relate directly to sequence variations at telomeres. In the present study, because C. hominis subtype IbA10G2 was shown to be more virulent than other subtypes in the study community (12), we compared population genetics of IbA10G2 and other gp60 subtypes. Heterogeneity within subtype IbA10G2 was observed in STRUCTURE and Network analyses compared with homogeneity within each of the remaining subtypes. This finding suggested a mixture of ancestral genetic elements and genetic recombination in virulent subtype IbA10G2. This finding was confirmed by incomplete LD and several recombination events (4 Rms) found in IbA10G2. In addition, the pattern of shared ancestral types in this study suggests that genetic exchange might have occurred between the ancestor of subtype IbA10G2 and the ancestor of subtype IaA13R8 or IeA11G3T3.
Pairwise FST analysis of 25 polymorphic loci in chromosome 6 between the virulent IbA10G2 subtype and non-IbA10G2 subtypes showed population differentiation at 4 loci around gp60. When a locus shows extraordinary levels of genetic population differentiation compared with other loci, this finding might be interpreted as evidence for positive selection (35). Thus, the region around gp60 in subtype IbA10G2 might be under selection pressure. This finding was further confirmed by comparison of haplotype diversity of the 2 groups at the 25 polymorphic loci. Although a similar Hd was found between the 2 groups in most regions of chromosome 6, a region of Hd reduction was observed in subtype IbA10G2 in a 129-kb region flanking gp60, compared with the non-IbA10G2 subtypes.
One explanation for this pattern of genetic diversity is that the region surrounding gp60 was probably affected by selective sweep or genetic hitchhiking caused by selection of the virulence subtype. A hallmark of a selective sweep is a chromosomal region with reduced diversity associated with a specific phenotype. In previous studies of other apicomplexan parasites, similar patterns of reduced genetic variations were observed in chromosomal regions surrounding sites under selection pressure. Wootton et al. (36) found a dramatic reduction in genetic variation in chloroquine-resistant parasites within a region spanning >200 kb around the Plasmodium falciparum chloroquine-resistance transporter gene in chromosome 7 as the result of selection for chloroquine resistance. Nair et al. (37) observed decreased variation in an ≈100-kb region flanking the dihydrofolate reductase gene in chromosome 4 of P. falciparum in association with pyrimethamine resistance. Consistent with these findings, we detected reduced sequence variation around gp60 in virulent subtype IbA10G2, suggesting that the 129-kb region surrounding gp60 in chromosome 6, perhaps gp60 itself, might be involved in selection for virulent gp60 subtype IbA10G2 in C. hominis. In a comparison of the number of nonsynonymous substitutions per nonsynonymous site to the number of synonymous substitutions per synonymous site analysis of loci around gp60, we confirmed the presence of positive section at least at the CP47 locus.
The reason for exclusive occurrence of genetic recombination in IbA10G2 is not clear. Because IbA10G2 is the major subtype of C. hominis responsible for numerous waterborne and foodborne outbreaks of cryptosporidiosis in many countries (2), it is transmitted frequently among humans, resulting in increased probability of mixed infections with other genetically heterogeneous subtypes, especially in countries in Europe in which autochthonous C. hominis infections are caused mostly by IbA10G2 and imported cases by other subtypes (38). In a long evolutionary process, the common occurrence and biologic fitness of the gp60 IbA10G2 subtype may facilitate genetic recombination with other C. hominis subtypes and subsequent spread of the recombinant parasite with the fitness gene. Recently, genetic recombination was shown to be a key strategy for selection of virulent clones of Toxoplasma gondii, an apicomplexan parasite with a largely clonal population structure in North America and Europe (39).
In conclusion, we have shown complex substructures in a natural C. hominis population in a cohort of children living in a small community in Peru. Although C. hominis from the community has an overall clonal population structure, genetic recombination occurs within subtype IbA10G2 around the gp60 locus, which might be involved in pathogenicity. Common occurrence of its parental subtypes and biologic fitness of the recombinant subtype with the IbA10G2 trait have probably facilitated genetic exchange and spread of the virulent subtype. In addition, we were able to localize selection for the virulent subtype IbA10G2 to a 129-kb region surrounding gp60 in chromosome 6. These observations could improve our understanding of emergence and spread of virulent C. hominis subtypes.
Acknowledgments
This study was supported in part by the National Natural Science Foundation of China (grants 31110103901 and 31229005), the National Basic Research Program of China (973 project no. 2011CB200903), the National Institutes of Health (grants 5P01AI051976 and 5R21AI059661), the US Department of Agriculture–Cooperative State Research, Education, and Extension Service (grant 2001-51110-11340), and Fundamental Research Funds for the Central Universities, China.
Dr Li is a lecturer at East China University of Science and Technology, Shanghai, China. Her research interests include molecular epidemiology and population genetics of zoonotic parasites.
References
- Chalmers RM, Davies AP. Minireview: clinical cryptosporidiosis. Exp Parasitol. 2010;124:138–46 . DOIPubMedGoogle Scholar
- Xiao L. Molecular epidemiology of cryptosporidiosis: an update. Exp Parasitol. 2010;124:80–9 . DOIPubMedGoogle Scholar
- Zhou L, Singh A, Jiang J, Xiao L. Molecular surveillance of Cryptosporidium spp. in raw wastewater in Milwaukee: implications for understanding outbreak occurrence and transmission dynamics. J Clin Microbiol. 2003;41:5254–7 . DOIPubMedGoogle Scholar
- Chalmers RM, Robinson G, Elwin K, Hadfield SJ, Thomas E, Watkins J, Detection of Cryptosporidium species and sources of contamination with Cryptosporidium hominis during a waterborne outbreak in north west Wales. J Water Health. 2010;8:311–25. DOIPubMedGoogle Scholar
- Mayne DJ, Ressler KA, Smith D, Hockey G, Botham SJ, Ferson MJ. A community outbreak of cyrptosporidiois in Sydney associated with a public swimming facility: a case–control study. Interdiscip Perspect Infect Dis. 2011;2011:341065. . Epub 2011 Dec 13. DOIGoogle Scholar
- Ng JS, Pingault N, Gibbs R, Koehler A, Ryan U. Molecular characterisation of Cryptosporidium outbreaks in Western and South Australia. Exp Parasitol. 2010;125:325–8. DOIPubMedGoogle Scholar
- Waldron LS, Ferrari BC, Cheung-Kwok-Sang C, Beggs PJ, Stephens N, Power ML. Molecular epidemiology and spatial distribution of a waterborne cryptosporidiosis outbreak in Australia. Appl Environ Microbiol. 2011;77:7766–71. DOIPubMedGoogle Scholar
- Cohen S, Dalle F, Gallay A, Di Palma M, Bonnin A, Ward HD. Identification of Cpgp40/15 type Ib as the predominant allele in isolates of Cryptosporidium spp. from a waterborne outbreak of gastroenteritis in South Burgundy, France. J Clin Microbiol. 2006;44:589–91. DOIPubMedGoogle Scholar
- Glaberman S, Moore JE, Lowery CJ, Chalmers RM, Sulaiman I, Elwin K, Three drinking-water–associated cryptosporidiosis outbreaks, Northern Ireland. Emerg Infect Dis. 2002;8:631–3. DOIPubMedGoogle Scholar
- Leoni F, Mallon ME, Smith HV, Tait A, McLauchlin J. Multilocus analysis of Cryptosporidium hominis and Cryptosporidium parvum isolates from sporadic and outbreak-related human cases and C. parvum isolates from sporadic livestock cases in the United Kingdom. J Clin Microbiol. 2007;45:3286–94. DOIPubMedGoogle Scholar
- Fournet N, Deege MP, Urbanus AT, Nichols G, Rosner BM, Chalmers RM, Simultaneous increase of Cryptosporidium infections in the Netherlands, the United Kingdom and Germany in late summer season, 2012. Euro Surveill. 2013;18:20348 .PubMedGoogle Scholar
- Cama VA, Bern C, Roberts J, Cabrera L, Sterling CR, Ortega Y, Cryptosporidium species and subtypes and clinical manifestations in children, Peru. Emerg Infect Dis. 2008;14:1567–74. DOIPubMedGoogle Scholar
- Bouzid M, Hunter PR, Chalmers RM, Tyler KM. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev. 2013;26:115–34. DOIPubMedGoogle Scholar
- Xiao L, Singh A, Limor J, Graczyk TK, Gradus S, Lal A. Molecular characterization of Cryptosporidium oocysts in samples of raw surface water and wastewater. Appl Environ Microbiol. 2001;67:1097–101. DOIPubMedGoogle Scholar
- Alves M, Xiao L, Sulaiman I, Lal AA, Matos O, Antunes F. Subgenotype analysis of Cryptosporidium isolates from humans, cattle, and zoo ruminants in Portugal. J Clin Microbiol. 2003;41:2744–7. DOIPubMedGoogle Scholar
- Cama VA, Arrowood MJ, Ortega YR, Xiao L. Molecular characterization of the Cryptosporidium parvum IOWA isolate kept in different laboratories. J Eukaryot Microbiol. 2006;53(Suppl 1):S40–2. DOIPubMedGoogle Scholar
- Gatei W, Das P, Dutta P, Sen A, Cama V, Lal AA, Multilocus sequence typing and genetic structure of Cryptosporidium hominis from children in Kolkata, India. Infect Genet Evol. 2007;7:197–205. DOIPubMedGoogle Scholar
- Gatei W, Hart CA, Gilman RH, Das P, Cama V, Xiao L. Development of a multilocus sequence typing tool for Cryptosporidium hominis. J Eukaryot Microbiol. 2006;53(Suppl 1):S43–8. DOIPubMedGoogle Scholar
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. DOIPubMedGoogle Scholar
- Kelly JK. A test of neutrality based on interlocus associations. Genetics. 1997;146:1197–206 .PubMedGoogle Scholar
- Vidigal PM, Mafra CL, Silva FM, Fietto JL, Silva Junior A, Almeida MR. Tripping over emerging pathogens around the world: a phylogeographical approach for determining the epidemiology of porcine circovirus-2 (PCV-2), considering global trading. Virus Res. 2012;163:320–7. DOIPubMedGoogle Scholar
- Widmer G. Population genetics of Cryptosporidium parvum. Trends Parasitol. 2004;20:3–6. DOIPubMedGoogle Scholar
- Feng X, Rich SM, Tzipori S, Widmer G. Experimental evidence for genetic recombination in the opportunistic pathogen Cryptosporidium parvum. Mol Biochem Parasitol. 2002;119:55–62. DOIPubMedGoogle Scholar
- Tanriverdi S, Blain JC, Deng B, Ferdig MT, Widmer G. Genetic crosses in the apicomplexan parasite Cryptosporidium parvum define recombination parameters. Mol Microbiol. 2007;63:1432–9. DOIPubMedGoogle Scholar
- Mallon M, MacLeod A, Wastling J, Smith H, Reilly B, Tait A. Population structures and the role of genetic exchange in the zoonotic pathogen Cryptosporidium parvum. J Mol Evol. 2003;56:407–17. DOIPubMedGoogle Scholar
- Mallon ME, MacLeod A, Wastling JM, Smith H, Tait A. Multilocus genotyping of Cryptosporidium parvum type 2: population genetics and sub-structuring. Infect Genet Evol. 2003;3:207–18. DOIPubMedGoogle Scholar
- Leav BA, Mackay MR, Anyanwu A. RM O’Connor RM, Cevallos AM, Kindra G, et al. Analysis of sequence diversity at the highly polymorphic Cpgp40/15 locus among Cryptosporidium isolates from human immunodeficiency virus–infected children in South Africa. Infect Immun. 2002;70:3881–90.
- Smith HV, Caccio SM, Cook N, Nichols RA, Tait A. Cryptosporidium and Giardia as foodborne zoonoses. Vet Parasitol. 2007;149:29–40. DOIPubMedGoogle Scholar
- Ngouanesavanh T, Guyot K, Certad G, Le Fichoux Y, Chartier C, Verdier RI, Cryptosporidium population genetics: evidence of clonality in isolates from France and Haiti. J Eukaryot Microbiol. 2006;53(Suppl 1):S33–6. DOIPubMedGoogle Scholar
- Peng MM, Meshnick SR, Cunliffe NA, Thindwa BD, Hart CA, Broadhead RL, Molecular epidemiology of cryptosporidiosis in children in Malawi. J Eukaryot Microbiol. 2003;50(Suppl):557–9. DOIPubMedGoogle Scholar
- Cevallos AM, Zhang X, Waldor MK, Jaison S, Zhou X, Tzipori S, Molecular cloning and expression of a gene encoding Cryptosporidium parvum glycoproteins gp40 and gp15. Infect Immun. 2000;68:4108–16. DOIPubMedGoogle Scholar
- Strong WB, Gut J, Nelson RG. Cloning and sequence analysis of a highly polymorphic Cryptosporidium parvum gene encoding a 60-kilodalton glycoprotein and characterization of its 15- and 45-kilodalton zoite surface antigen products. Infect Immun. 2000;68:4117–34. DOIPubMedGoogle Scholar
- Bouzid M, Hunter PR, McDonald V, Elwin K, Chalmers RM, Tyler KM. A new heterogeneous family of telomerically encoded Cryptosporidium proteins. Evol Appl. 2013;6:207–17.
- Widmer G, Lee Y, Hunt P, Martinelli A, Tolkoff M, Bodi K. Comparative genome analysis of two Cryptosporidium parvum isolates with different host range. Infect Genet Evol. 2012;12:1213–21. DOIPubMedGoogle Scholar
- Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218. DOIPubMedGoogle Scholar
- Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–3. DOIPubMedGoogle Scholar
- Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol. 2003;20:1526–36. DOIPubMedGoogle Scholar
- Chalmers RM, Hadfield SJ, Jackson CJ, Elwin K, Xiao L, Hunter P. Geographic linkage and variation in Cryptosporidium hominis. Emerg Infect Dis. 2008;14:496–8. DOIPubMedGoogle Scholar
- Herrmann DC, Barwald A, Maksimov A, Pantchev N, Vrhovec MG, Conraths FJ, Toxoplasma gondii sexual cross in a single naturally infected feline host: generation of highly mouse-virulent and avirulent clones, genotypically different from clonal types I, II and III. Vet Res. 2012;43:39. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 19, Number 10—October 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Yaoyu Feng, State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, China
Top