Volume 20, Number 4—April 2014
Research
Novel Betacoronavirus in Dromedaries of the Middle East, 2013
Cite This Article
Citation for Media
Abstract
In 2013, a novel betacoronavirus was identified in fecal samples from dromedaries in Dubai, United Arab Emirates. Antibodies against the recombinant nucleocapsid protein of the virus, which we named dromedary camel coronavirus (DcCoV) UAE-HKU23, were detected in 52% of 59 dromedary serum samples tested. In an analysis of 3 complete DcCoV UAE-HKU23 genomes, we identified the virus as a betacoronavirus in lineage A1. The DcCoV UAE-HKU23 genome has G+C contents; a general preference for G/C in the third position of codons; a cleavage site for spike protein; and a membrane protein of similar length to that of other betacoronavirus A1 members, to which DcCoV UAE-HKU23 is phylogenetically closely related. Along with this coronavirus, viruses of at least 8 other families have been found to infect camels. Because camels have a close association with humans, continuous surveillance should be conducted to understand the potential for virus emergence in camels and for virus transmission to humans.
The 2003 epidemic of severe acute respiratory syndrome (SARS) boosted interest in the discovery of new coronaviruses (CoVs) (1–3). In 2004, a novel human CoV (HCoV), named HCoV-NL63, was reported (4), and the discovery of another novel HCoV, HCoV-HKU1, was described and further characterized in 2005 (5,6) and 2006 (7). SARS-CoV–like viruses have also been reported in Chinese horseshoe bats in Hong Kong, China, and other horseshoe bats in China (8,9). The discovery in Chinese horseshoe bats in Yunnan, China, of a new SARS-CoV–like virus that uses ACE2 as receptor has furthered interest in discovering animal origins of human infections (10). We have discovered 20 other animal CoVs that include 2 novel betacoronavirus lineages and a novel genus, Deltacoronavirus (11–20). From our studies, bats and birds were shown to be the gene sources for fueling the evolution and dissemination of alphacoronaviruses and betacoronaviruses and of gammacoronaviruses and deltacoronaviruses, respectively (18).
In 2012, a novel CoV, Middle East respiratory syndrome CoV (MERS-CoV) emerged as a cause of severe respiratory infections associated with high rates of death among humans; the virus is closely related to tylonycteris bat CoV HKU4 and pipistrellus bat CoV HKU5 (Pi-Bat CoV HKU5) (21–23). It has also been shown that dromedaries in the Middle East possess MERS-CoV neutralizing antibodies (24). To further knowledge of the evolution and dissemination of CoVs, we conducted a molecular epidemiology study of fecal samples obtained from dromedaries in Dubai, United Arab Emirates.
Samples
Dromedary fecal and serum samples used in the study were leftover specimens that had been submitted for pathogen screening (feces) or preventive health screening (serum) to Central Veterinary Research Laboratory (Dubai, United Arab Emirates) during January–July 2013. The fecal and serum samples were not obtained from the same animals. None of the dromedaries tested were known to have had contact with bats or horses.
We tested a total of 293 fecal samples: 232 from teenage and adult dromedaries (Camelus dromedarius) (>1 year of age) and 61 from dromedary calves (<1 year of age). Among the 293 samples, 6 were collected in January, 209 in February, 5 in March, 39 in April, 16 in May, 7 in June, and 11 in July 2013.
We tested a total of 59 serum samples: 55 from teenage and 4 from adult dromedaries. The serum samples were collected from female dairy farm or racing dromedaries. The dairy dromedaries were purchased from various countries (e.g., Saudi Arabia, Oman, Sudan, and Pakistan); the number of dairy dromedaries from each country and their lengths of stay in Dubai were not known. The racing dromedaries were from Dubai Emirate.
RNA Extraction, Reverse Transcription PCR, and DNA Sequencing
Viral RNA extraction was conducted as described (20,23). Initial CoV screening was performed by amplifying a 440-bp fragment of CoV RNA-dependent RNA polymerase (RdRp) gene by using conserved primers (5′-GGTTGGGACTATCCTAAGTGTGA-3′ and 5′-ACCATCATCNGANARDATCATNA-3′ and degenerative bases N, R, and D, representing A/C/T/G, A/G, and A/G/T, respectively. After the novel CoV was detected in samples, subsequent screening was performed by using specific primers 5′-ACTATGACTGGCAGAATGTT-3′ and 5′-TAATAAGGCGACGTAACATA-3′. To amplify a 126-bp fragment of RdRp gene, reverse transcription PCR (RT-PCR) and DNA sequencing were performed as described (20,23).
Virus Culture
The fecal samples from 3 dromedaries tested positive for CoV. These samples were cultured in HRT-18G, Vero E6, Caco-2, and LLC-MK2 cell lines.
Complete Genome Sequencing and Analysis
Three complete genomes of DcCoV UAE-HKU23 (265F, 362F, and 368F) were amplified and sequenced as described (5,12). RNA extracted from fecal specimens was used as template, and a database of CoV genes and genomes (CoVDB, http://covdb.microbiology.hku.hk) (25) was used for sequence retrieval. Sequences were assembled and edited to produce final sequences.
We used EMBOSS Needle (http://www.ebi.ac.uk/Tools/psa/emboss_needle/) to compare the nucleotide sequences of the genomes and the deduced amino acid sequences with those for other CoVs. A neighbor-joining phylogenetic tree with 1,000 bootstraps was constructed by using the Jones-Taylor-Thornton substitution model; gamma distribution among sites was conducted in MEGA5 (26).
Real-time Quantitative RT-PCR
According to our protocol (19,20), all samples positive for DcCoV UAE-HKU23 by RT-PCR were subjected to quantitative RT-PCR (qRT-PCR).The assay was performed, as described (27), using a real-time 1-step qRT-PCR with DcCoV UAE-HKU23 primers 5′-ATAGCGGCTACACGTGGTGTT-3′ and 5′-TCCCAGCCGCCATAAAACT-3′ and probe 5′-(FAM) CTGTTGTTATAGGCACCACT (BHQ1)-3′. To generate calibration curves, we prepared a series of 6 log10 dilutions equivalent to 101–106 copies per reaction mixture and ran them in parallel with the test samples.
Cloning and Purification
The nucleocapsid proteins of SARS-CoV (betacoronavirus lineage B), Pi-Bat CoV HKU5 (betacoronavirus lineage C), and rousettus bat coronavirus (Ro-BatCov HKU9; betacoronavirus lineage D) were obtained as described (2,16). To produce a plasmid for DcCoV UAE-HKU23 nucleocapsid protein purification, primers 5′-CATGCCATGGGCATGTCTTTTACTCCTGGTAAGC-3′ and 5′-CCGCTCGAGTATTTCTGAGGTGTTTTCAG-3′ were used to amplify the gene encoding the nucleocapsid protein of DcCoV UAE-HKU23. The sequence encoding aa 1–672 of the nucleocapsid protein was amplified and cloned into the NcoI and XhoI sites of expression vector pET-28b(+) (Merck KGaA, Darmstadt, Germany). Recombinant nucleocapsid protein was expressed and purified by using the Ni2+-loaded HiTrap Chelating System (GE Healthcare Life Sciences, Little Chalfont, UK) according to the manufacturer's instructions. The nucleocapsid protein of MERS-CoV was cloned and purified by using the method described above with primers 5′-GGAATTCCATATGATGGCATCCCCTGCTGCACCTC-3′ and 5′-ATAAGAATGCGGCCGCATCAGTGTTAACATCAATCATT-3′.
Western Blot Analysis
Western blot analysis was performed as described (5) with 1.5 I1/4g of purified (His)6-tagged recombinant nucleocapsid protein of DcCoV UAE-HKU23 and 1:2,000, 1:4,000, and 1:8,000 dilutions of dromedary serum samples. Antigen–antibody interaction was detected by using 1:4,000 diluted horseradish peroxidase–conjugated Goat Anti-Llama IgG (Life Technologies, Carlsbad, CA, USA) and the ECL fluorescence system (GE Healthcare Life Sciences).
To determine the possibility of cross-reactivity between antibodies against the nucleocapsid protein of DcCoV UAE-HKU23 and that of other betacoronavirus lineages, we tested 3 serum samples that were positive for antibody against the nucleocapsid protein of DcCoV UAE-HKU23. We used 1.5 I1/4g of purified (His)6-tagged recombinant nucleocapsid protein of 3 betacoronaviruses (SARS-CoV [lineage B], Pi-BatCoV HKU5 [lineage C], and Ro-BatCoV HKU9 [lineage D]) and 1:2,000 dilutions of serum samples. To determine the presence of antibodies against the nucleocapsid protein of MERS-CoV, we tested the serum samples by using 1.5 I1/4g of purified (His)6-tagged recombinant nucleocapsid protein of MERS-CoV and 1:2,000 dilutions of serum samples. Antigen–antibody interaction was detected as described above.
Indirect Immunofluorescence
Anti–MERS-CoV antibody detection by indirect immunofluorescence was performed as described (28) with minor modifications. In brief, Vero cells infected with MERS-CoV were prepared as described (28). Camel serum samples were screened at a dilution of 1:160 on infected and noninfected control cells. Antigen–antibody interaction was detected by using fluorescein isothiocyanate–conjugated Goat Anti-Llama IgG (Life Technologies). Serum samples positive at a screening dilution of 1:160 were further titrated with serial 2-fold dilutions. The indirect immunofluorescence antibody titer was the highest dilution giving a positive result.
Neutralization Antibody Test
The neutralization antibody test was performed as described (28). In brief, starting with a serum dilution of 1:10, we prepared serial 2-fold dilutions of serum in 96-well microtiter plates. For each serum dilution, 0.05 mL of the dilution was mixed with 0.05 mL of 200 MERS-CoV 50% tissue culture infectious doses and incubated at 37°C for 1.5 h in a CO2 incubator. Then 0.1 mL of virus–serum mixture was inoculated in duplicate wells of 96-well microtiter plates with preformed monolayers of Vero cells and further incubated at 37°C for 3–4 days. Cytopathic effects were observed by using an inverted microscope on days 3 and 4 after inoculation. The neutralizing antibody titer was determined as the highest dilution of serum that completely suppressed the cytopathic effects in at least half of the infected wells.
Estimation of Substitution Rates and Divergence Dates
The number of synonymous substitutions per synonymous site, Ks, and the number of nonsynonymous substitutions per nonsynonymous site, Ka, for each coding region between each pair of strains were calculated by using the Nei-Gojobori method (Jukes-Cantor) in MEGA5 (26). Divergence time was calculated on the basis of RdRp gene sequence data by using a Bayesian Markov Chain Monte Carlo approach as implemented in BEAST version 1.8.0 (http://beast.bio.ed.ac.uk), as described (15,19,29,30). Bayesian skyline under a relaxed-clock model with an uncorrelated exponential distribution was adopted for making inferences because Bayes factor analysis for the RdRp gene indicated that this model fitted the data better than other models tested.
Identification of CoV in Dromedaries
Of the 293 fecal samples tested, 14 (4.8%) were RT-PCR positive for the CoV RdRp gene; 1 (0.4%) of the samples was from an adult dromedary and 13 (21.3%) were from calves (Table 1). Of the 14 positive samples, 11 were collected in April and 3 were collected in May. Ten of the 14 samples were submitted to the Central Veterinary Research Laboratory for routine checking, and the other 4 were collected because the dromedaries had diarrhea. Sequencing results indicated a 126-nt sequence identical to that of equine CoV, a betacoronavirus 1 species in lineage A (betacoronavirus A1).
Virus Culture and Virus Load
Attempts to stably passage DcCoV UAE-HKU23 in cell cultures were unsuccessful; no cytopathic effect or viral replication was detected. Real-time qRT-PCR showed that the amounts of DcCoV UAE-HKU23 RNA ranged from 5.7 A- 104 copies/mL to 9.8 A- 107 copies/mL (median 8.4 A- 105) in the 14 fecal samples positive for DcCoV UAE-HKU23 (Table 1).
Genome Organization and Coding Potential
Figure 1

Figure 1. . . . . . Genome organizations of a novel betacoronavirus, in boldface, discovered in dromedaries in the Middle East in 2013, and representative coronaviruses from each coronavirus genus (labeled on...
The 3 complete genomes of DcCoV UAE-HKU23 (GenBank accession nos. KF906249–KF906251) were 31,036 bases and had a G+C content of 37% (Table 2). The genome organization is similar to that of other betacoronavirus lineage A CoVs (Figure 1). Additional open-reading frames (ORFs) coding for nonstructural proteins (NSPs) NS2 and NS5 are found. DcCoV UAE-HKU23 and other CoVs in betacoronavirus lineage A possess the same putative transcription regulatory sequence motif, 5′-UCUAAAC-3′, at the 3′ end of the leader sequence and preceding most ORFs (Table 3; Technical Appendix Table 1) (19,30).
The characteristics of putative NSPs in ORF1ab of DcCoV UAE-HKU23 are shown in Technical Appendix Table 2. The ORF1ab polyprotein shared 70.7%–99.3% aa identity with polyproteins of other betacoronavirus lineage A CoVs. The predicted putative cleavage sites were conserved between DcCoV UAE-HKU23 and other members of betacoronavirus A1 of the Betacoronavirus genus. The lengths of NSPs 1–3, 13, and 15 in DcCoV UAE-HKU23 differed from those in equine CoV, porcine hemagglutinating encephalomyelitis virus, and/or HCoV-OC43 as a result of deletions/insertions.
Figure 2

Figure 2. . Amino acid comparison of the spike protein of a novel betacoronavirus, dromedary camel coronavirus (DcCoV) UAE-HKU23, discovered in dromedaries in the Middle East in 2013, with that of bovine coronavirus...
The amino acid sequence of the predicted spike protein of DcCoV UAE-HKU23 is most similar to that of bovine coronavirus (BCoV) and sable antelope CoV, with which DcCoV UAE-HKU23 has 94.1% similarity (Table 2). A comparison of the amino acid sequences of DcCoV UAE-HKU23 spike protein and BCoV spike protein showed 81 aa polymorphisms, of which 24 were seen within the region previously identified as hypervariable among the spike protein of other betacoronavirus lineage A CoVs (31) (Figure 2); this finding suggests that this region in DcCoV UAE-HKU23 is also subject to strong immune selection. BCoV has been found to use N-acetyl-9-O acetyl neuramic acid as a receptor for initiation of infection (32). Among the 5 aa that may affect S1-mediated receptor binding in BCoV (31), 2 aa (threonine at position 11 and glutamine at position 179) were conserved in DcCoV UAE-HKU23 (Figure 2). However, at positions 115, 118, and 173, aspartic acid, methionine, and asparagine observed in BCoV and were replaced by serine, threonine, and histidine, respectively, in DcCoV UAE-HKU23. A recent report identified 4 aa acids that were critical sugar-binding residues in the spike protein of BCoV (tyrosine, glutamic acid, tryptophan, and histidine at positions 162, 182, 184, and 185, respectively) (32); all 4 aa were also present in spike protein of DcCoV UAE-HKU23 (Figure 2). Another study identified 7 aa substitutions in the spike protein of BCoV that differed between virulent and avirulent, cell culture-adapted strains (31); 5 of the 7 aa from virulent strains were also conserved in DcCoV UAE-HKU23, and amino acid substitutions were observed in the other 2 aa (threonine→valine at position 40 and aspartic acid→asparagine at position 470). It has also been reported that an amino acid change at position 531 of the spike protein of BCoV discriminated between enteric (aspartic acid/asparagine) and respiratory (glycine) strains (33). At this position, a threonine was conserved in all 3 genomes of DcCoV UAE-HKU23.
NS5 of DcCoV UAE-HKU23 shares 83.5%–98.2% aa identity with the corresponding NSPs of betacoronavirus A1 members. In murine hepatitis virus, translation of the envelope protein is cap-independent, through an internal ribosomal entry site (19,30). However, a preceding transcription regulatory sequence, 5′-UCCAAAC-3′, can be identified upstream of the envelope protein in DcCoV UAE-HKU23, as in other betacoronavirus A1 members (Table 3; Technical Appendix Table 1) (19,30). Downstream to nucleocapsid protein, the 3′-untranslated region contains a predicted bulged stem-loop structure of 68 nt (nt position 30747–30814) that is conserved in betacoronaviruses (34). Overlapping with the bulged stem-loop structure by 5 nt, is a conserved pseudoknot structure (nt position 30810–30863) that is important for CoV replication.
Phylogenetic Analyses
Figure 3

Figure 3. . Phylogenetic analysis of open reading frame (ORF) 1b polyprotein of dromedary camel coronavirus (DcCoV) UAE-HKU23 from dromedaries of the Middle East, 2013. The tree was constructed by the neighbor-joining method,...
Figure 5
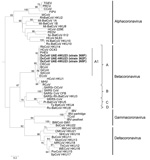
Figure 5. . . . . . Phylogenetic analyses of the nucleocapsid protein of a novel coronavirus (CoV), dromedary camel CoV (DcCoV) UAE-HKU23, discovered in dromedaries of the Middle East, 2013.The tree was...
Phylogenetic trees constructed by using the amino acid sequences of ORF1b polyprotein, spike protein, and nucleocapsid protein of DcCoV UAE-HKU23 and other CoVs are shown in Figures 3–5. The pairwise amino acid identities of chymotrypsin-like protease (3CLpro), RdRp, helicase, spike protein, and nucleocapsid protein are shown in Table 2. In all 3 phylogenetic trees, DcCoV UAE-HKU23 clustered with other members of betacoronavirus A1, including BCoV, sable antelope CoV, equine CoV, HCoV-OC43, giraffe CoV, porcine hemagglutinating encephalomyelitis virus, canine respiratory CoV, and rabbit CoV (RbCoV) HKU14 (Figures 3–5).
Antibody Detection
Figure 6
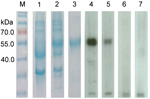
Figure 6. . . . . . Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blot analysis of a novel coronavirus, dromedary camel coronavirus UAE-HKU23, discovered in dromedaries of the Middle East, 2013.Nucleocapsid protein...
Nucleocapsid protein of DcCoV UAE-HKU23 was purified. Prominent immunoreactive bands were visible for 31 (52%) of 59 dromedary serum samples; 25 of the 31 samples had titers of 2,000, three had titers of 4,000, and 3 had titers of 8,000 (Figure 6). Serum samples for all 4 adult dromedaries were positive for DcCoV UAE-HKU23 antibodies, and 27 (49%) of the 55 samples for teenage dromedaries were positive. Band sizes were ≈50 kDa, consistent with the expected size of 50.4 kDa for the full-length (His)6-tagged recombinant nucleocapsid protein. Only very faint bands were observed when the 3 serum samples positive for DcCoV UAE-HKU23 antibodies were incubated with nucleocapsid proteins of SARS-CoV, Pi-BatCoV HKU5, or Ro-BatCoV HKU9, indicating minimal cross-reactivity. This finding concurs with our previous observation that minimal cross-reactivity occurs between CoVs in different lineages in betacoronavirus (16). For MERS-CoV antibody testing, results were positive for 57 (97%) of the 59 samples by Western blot analysis, for all 59 samples by indirect immunofluorescence, and for 58 (98%) of the 59 samples by neutralization antibody test (Table 4).
Estimation of Substitution Rates and Divergence Dates
The Ka, Ks, and Ka/Ks of the various coding regions in DcCoV UAE-HKU23 are shown in Table 5. The Ka/Ks of all the coding regions in DcCoV UAE-HKU23 was <0.5.
By using the uncorrelated relaxed clock model on RdRp gene sequences, we estimated the date of divergence between DcCoV UAE-HKU23 and BCoV to be ≈46 years ago. We estimated that the 3 strains of DcCoV UAE-HKU23 diverged from their most recent common ancestor in March 2010 (the 95% highest posterior density interval, August 2006–September 2012) (Technical Appendix Figure 1).
We discovered a novel CoV, but no MERS-CoV, in dromedaries from the Middle East. Dromedaries are 1 of 2 surviving camel species. Dromedaries (C. dromedarius; 1-humped camels) inhabit the Middle East and northern and northeastern Africa; Bactrian camels (C. bactrianus, 2-humped camels) inhabit Central Asia. Among the 20 million camels on earth, 90% are dromedaries. In 2012, there were ≈360,000 dromedaries in the United Arab Emirates.
In this study, we discovered a novel CoV, DcCoV UAE-HKU23, from 4.8% of 293 fecal samples collected from dromedaries in Dubai. The positive samples were not collected from the same farm or stable. Moreover, there was >0.2% nt difference among the 3 complete genomes sequenced, indicating that the positive samples were not collected from a clonal outbreak. In our study, 21.3% of dromedary calves, but only 0.4% of adult dromedaries, were RT-PCR positive for DcCoV UAE-HKU23; this finding indicates that dromedary calves are probably more susceptible than adult dromedaries to infection with DcCoV UAE-HKU23. Furthermore, DcCoV UAE-HKU23 is probably stably evolving in dromedaries because the Ka/Ks of all the coding regions in the genome were <0.5. In this study, 4 of the 12 positive samples were collected from dromedaries with diarrhea. A previous report also described the presence of a betacoronavirus in the fecal sample of a dromedary calf with diarrhea (35). This finding raises the question of the pathologic significance of DcCoV UAE-HKU23 for camelids and warrants further animal studies.
Our serologic data showed little cross-reactivity between DcCoV UAE-HKU23 and SARS-CoV, Pi-BatCoV HKU5, and Ro-BatCoV HKU9. This finding is in line with findings from our previous studies of Ro-BatCoV HKU9, which also showed minimal serologic cross-reactivity among the 4 lineages of betacoronaviruses (16). These results suggest that there should be minimal cross-reactivity between DcCoV UAE-HKU23 and MERS-CoV, which belong to 2 different CoV lineages. Because we showed an extremely high prevalence of MERS-CoV antibodies in the serum samples by Western blot analysis, indirect immunofluorescence, and neutralization antibody testing, concurring with findings in a previous study (24), we would also expect a similar high prevalence of DcCoV UAE-HKU23 antibodies if there was major serologic cross-reactivity between MERS-CoV and DcCoV UAE-HKU23. However, our serologic data only revealed the presence of DcCoV UAE-HKU23 antibodies in 52% of the serum samples, indicating that no correlation exists between seropositivity to DcCoV UAE-HKU23 and seropositivity to MERS-CoV. Furthermore, we found no correlation between seropositivity to DcCoV UAE-HKU23 and MERS-CoV antibody titers.
In this study, correlation between DcCoV UAE-HKU23 RT-PCR positivity and seropositivity also cannot be ascertained because the fecal samples and serum samples were collected from different dromedaries. Because MERS-CoV was not present in dromedaries in the present study, an intensive search in dromedaries and other animals in other locations in the Middle East would be helpful in the search for the animal source of MERS-CoV.
Figure 7
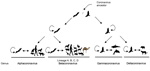
Figure 7. . . . . . The evolution of corona viruses from their ancestors in bat and bird hosts to new virus species that infect other animals.
DcCoV UAE-HKU23 is a member of betacoronavirus A1 (Figure 7). Comparison of the amino acid identities of the 7 conserved replicase domains for species demarcation (i.e., ADP-ribose 1″-phosphatase, NSP5 [3CLpro], NSP12 [RdRp], NSP13 [helicase], NSP14 [ExoN], NSP15 [NendoU], and NSP16 [O-MT]) (36) between DcCoV UAE-HKU23 and other CoVs of betacoronavirus A1 revealed that in all 7 domains, the amino acid sequences of DcCoV UAE-HKU23 and other betacoronavirus A1 members shared >90% identity. This finding indicates that DcCoV UAE-HKU23 should be a member of betacoronavirus A1.
Furthermore, the genome characteristics of DcCoV UAE-HKU23 showed features similar to those of other betacoronavirus A1 members. The genomes of all betacoronavirus A1 members have G+C contents of 0.37 and the genome of RbCoV HKU14, a recently discovered CoV closely related to betacoronavirus A1 (19), has a G+C content of 0.38. This G+C content differs substantially from those of other CoV species of lineage A betacoronaviruses, which have G+C contents of 0.32 (HCoV-HKU1), 0.41 (rat CoV) and 0.42 (murine hepatitis virus) (Table 2). The difference in the genome characteristics between A1 and non-A1 members of betacoronavirus is also reflected by their codon usage bias, in which the general preference of using G/C in the third position of the codons decreases from murine hepatitis virus and rat CoV to betacoronavirus A1 members and RbCoV HKU14 to HCoV-HKU1 (Technical Appendix Figure 2). The cleavage site for spike protein of betacoronavirus A1 members is RRS/QRR, whereas those of HCoV-HKU1, RbCoV HKU14, and murine hepatitis virus are RRKRR, LRSRR, and RAR/H/DR/S, respectively. The length of membrane genes for betacoronavirus A1 members and RbCoV HKU14 is 693 bases, whereas the lengths for HCoV-HKU1 and murine hepatitis virus are 672 and 687 bases, respectively.
DcCoV UAE-HKU23 is phylogenetically closely related to other betacoronavirus A1 members, which in turn are closely related to other CoVs of betacoronavirus lineage A. Despite their close relationships, no recombination was detected between DcCoV UAE-HKU23 and other betacoronavirus A1 members by bootscan analysis (data not shown). These CoVs of betacoronavirus A1 may be using different receptors in their corresponding hosts because their spike protein is one of the proteins that show the largest difference among different CoVs. Most of the differences among the spike proteins in different betacoronavirus A1 members were also observed in the N terminal half of their spike protein, where the receptor binding domains should be located.
Camels are one of the most unique mammals on earth. In particular, they have shown perfect adaptation to desert life, which presents temperature extremes and a scarce supply of food and water. In the past, camels were used for transportation of humans and goods and for military uses. Moreover, for humans, they provide a good source of meat, milk, and wool. Camels are also important recreational animals in the Middle East and are used for camel racing. Having been associated with humans for at least 5,000 years, camels usually pose little physical danger to humans. However, infectious pathogens, such as brucellosis, can occasionally be transmitted from camels to humans. Apart from the present novel CoV, viruses of at least 8 taxonomic families (i.e., Paramyxoviridae, Flaviviridae, Herpesviridae, Papillomaviridae, Picornaviridae, Poxviridae, Reoviridae, and Rhabdoviridae) have been found to infect camels (37–39). Because camels are closely associated with humans, continuous surveillance of viruses in this hardy group of animals is needed to understand the potential for virus emergence and transmission to humans.
Acknowledgments
We thank Wing-Man Ko and Constance Chan for their continuous support.
This work is partly supported by the Hong Kong Special Administrative Region Health and Medical Research Fund; Seed Funding for TRS and Strategic Research Theme Fund, The University of Hong Kong; Theme-based Research Scheme, Research Grant Council Grant, University Grant Council; and Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease for the Hong Kong Special Administrative Region Department of Health.
Dr Woo is a professor and head of the Department of Microbiology at The University of Hong Kong. His research focuses on novel microbe discovery and microbial genomics.
References
- Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–8. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Tsoi HW, Chan KH, Wong BH, Che XY, Relative rates of non-pneumonic SARS coronavirus infection and SARS coronavirus pneumonia. Lancet. 2004;363:841–5. DOIPubMedGoogle Scholar
- Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20:660–94. DOIPubMedGoogle Scholar
- van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Identification of a new human coronavirus. Nat Med. 2004;10:368–73. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79:884–95. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Tsoi HW, Huang Y, Poon RW, Chu CM, Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J Infect Dis. 2005;192:1898–907. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Yip CC, Tse H, Tsoi HW, Cheng VC, Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol. 2006;44:2063–71. DOIPubMedGoogle Scholar
- Lau SK, Woo C, Li KS, Huang Y, Tsoi HW, Wong BH, Severe acute respiratory syndrome coronavirus–like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–5. DOIPubMedGoogle Scholar
- Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–9. DOIPubMedGoogle Scholar
- Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, solation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–8. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Li S, Poon RW, Wong BH, Tsoi HW, Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–7. DOIPubMedGoogle Scholar
- Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol. 2007;81:1574–85. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Li KS, Huang Y, Wang M, Lam CS, Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology. 2007;367:428–39. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Lam CS, Lai KK, Huang Y, Lee P, Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J Virol. 2009;83:908–17. DOIPubMedGoogle Scholar
- Lau SK, LiK S, Huang Y, Shek CT, Tse H, Wang M, et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome–related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol. 2010;84:2808–19. DOIPubMedGoogle Scholar
- Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J Virol. 2010;84:11385–94. DOIPubMedGoogle Scholar
- Lau SK, Li KS, Tsang AK, Shek CT, Wang M, Choi GK, Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault's rousettes to pomona leaf-nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J Virol. 2012;86:11906–18. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol. 2012;86:3995–4008. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Yip CC, Fan RY, Huang Y, Wang M, Isolation and characterization of a novel Betacoronavirus subgroup A coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J Virol. 2012;86:5481–96. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Lam CS, Tsang AK, Hui SW, Fan RY, Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. J Virol. 2014;88:1318–31. DOIPubMedGoogle Scholar
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–20. DOIPubMedGoogle Scholar
- de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J Virol. 2013;87:7790–2. DOIPubMedGoogle Scholar
- Lau SK, Li KS, Tsang AK, Lam CS, Ahmed S, Chen H, Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of Pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J Virol. 2013;87:8638–50. DOIPubMedGoogle Scholar
- Reusken CB, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, Meyer B, Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis. 2013;13:859–66. DOIPubMedGoogle Scholar
- Huang Y, Lau SK, Woo PC, Yuen KY. CoVDB: a comprehensive database for comparative analysis of coronavirus genes and genomes. Nucleic Acids Res. 2008;36:D504–11. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Lau SK, Lau CC, Chan KH, Li CP, Chen H, Jin DY, Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. 2013;94:2679–90. DOIPubMedGoogle Scholar
- Chan KH, Chan JF, Tse H, Chen H, Lau CC, Cai JP, Cross-reactive antibodies in convalescent SARS patients' sera against the emerging novel human coronavirus EMC (2012) by both immunofluorescent and neutralizing antibody tests. J Infect. 2013;67:130–40. DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- Lau SK, Lee P, Tsang AK, Yip CC, Tse H, Lee RA, Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol. 2011;85:11325–37. DOIPubMedGoogle Scholar
- Chouljenko VN, Kousoulas KG, Lin X, Storz J. Nucleotide and predicted amino acid sequences of all genes encoded by the 3′ genomic portion (9.5 kb) of respiratory bovine coronaviruses and comparisons among respiratory and enteric coronaviruses. Virus Genes. 1998;17:33–42. DOIPubMedGoogle Scholar
- Peng G, Xu L, Lin YL, Chen L, Pasquarella JR, Holmes KV, Crystal structure of bovine coronavirus spike protein lectin domain. J Biol Chem. 2012;287:41931–8. DOIPubMedGoogle Scholar
- Yoo D, Deregt D. A single amino acid change within antigenic domain II of the spike protein of bovine coronavirus confers resistance to virus neutralization. Clin Diagn Lab Immunol. 2001;8:297–302.PubMedGoogle Scholar
- Goebel SJ, Hsue B, Dombrowski TF, Masters PS. Characterization of the RNA components of a putative molecular switch in the 3′ untranslated region of the murine coronavirus genome. J Virol. 2004;78:669–82. DOIPubMedGoogle Scholar
- WA1/4nschmann A, Frank R, Pomeroy K, Kapil S. Enteric coronavirus infection in a juvenile dromedary (Camelus dromedarius). J Vet Diagn Invest. 2002;14:441–4. DOIPubMedGoogle Scholar
- de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya A, Holmes KV, Coronaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy, classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Academic Press; 2011. pp. 806–28.
- Wernery U, Zachariah R. Experimental camelpox infection in vaccinated and unvaccinated dromedaries. Zentralbl Veterinarmed B. 1999;46:131–5.PubMedGoogle Scholar
- Wernery U, Kaaden OR. Foot-and-mouth disease in camelids: a review. Vet J. 2004;168:134–42. DOIPubMedGoogle Scholar
- Wernery U, Knowles NJ, Hamblin C, Wernery R, Joseph S, Kinne J, Abortions in dromedaries (Camelus dromedarius) caused by equine rhinitis A virus. J Gen Virol. 2008;89:660–6. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 20, Number 4—April 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Patrick CY Woo, State Key Laboratory of Emerging Infectious Diseases, Department of Microbiology, University of Hong Kong, University Pathology Bldg, Queen Mary Hospital, Hong Kong, ChinaPatrick CY Woo, State Key Laboratory of Emerging Infectious Diseases, Department of Microbiology, University of Hong Kong, University Pathology Bldg, Queen Mary Hospital, Hong Kong, China
Top