Volume 20, Number 6—June 2014
Dispatch
New Hepatitis E Virus Genotype in Camels, the Middle East
Cite This Article
Citation for Media
Abstract
In a molecular epidemiology study of hepatitis E virus (HEV) in dromedaries in Dubai, United Arab Emirates, HEV was detected in fecal samples from 3 camels. Complete genome sequencing of 2 strains showed >20% overall nucleotide difference to known HEVs. Comparative genomic and phylogenetic analyses revealed a previously unrecognized HEV genotype.
Hepatitis E virus (HEV) belongs to the family Hepeviridae and genus Hepevirus. Among humans worldwide, HEV is the most common cause of acute viral hepatitis. The disease is generally self-limiting, but mortality rates are high among pregnant women and young infants. Chronic HEV infection is a problem for immunocompromised patients, such as those who have received a solid organ transplant and those with HIV infection. In addition to humans, HEV has been found in the other mammals: pigs, boar, deer, rodents, ferrets, rabbits, mongoose, bats, cattle, sheep, foxes, minks, and horses (1–3). Among the 4 known HEV genotypes, HEV1 and HEV2 infect only humans; whereas, HEV3 and HEV4 can infect humans, pigs, and other mammals. Human infections with HEV3 and HEV4 have been associated with consumption of raw or undercooked pork or game meat (4). Traditionally, HEV infection is mainly transmitted through water contaminated with infected feces. Since water supplies and sanitary infrastructures have been improved, animals have become a major source of human HEV infection. We detected HEV in fecal samples from dromedary camels in the Middle East.
As part of a molecular epidemiology study, 203 fecal samples from 203 adult dromedaries (Camelus dromedarius) were submitted to the Central Veterinary Research Laboratory in Dubai, United Arab Emirates, over a 7-month period (January–July 2013). RNA extraction and reverse transcription were performed, as described, to detect other positive-sense single-stranded RNA viruses (5,6). Screening for HEV was performed by PCR amplification of a 284-bp fragment of open reading frame (ORF) 2 in HEV; specific primers used were 5′-TTTATTCTCGTCCAGTCGTTTC-3′ and 5′-GTCAGTGGAGGACCCATATGT-3′, designed from sequence information from our metagenomic study (P.C.Y. Woo et al., unpub. data). PCR was performed according to previously described conditions (7); annealing temperature were set at 50°C. DNA sequencing and quantitative real-time reverse transcription PCR were also performed as described (8). Using strategies we have reported for other positive-sense single-stranded RNA viruses, we performed complete-genome sequencing on 2 HEV-positive samples (5,6). Comparative genomic analysis was performed as described (9). Phylogenetic analysis was conducted in MrBayes5D version 3.1.2 (www.fifthdimension.jp/products/mrbayes5d/) by using an optimal substitution model with 1 million Markov chain Monte Carlo generations; sampling was conducted every 100 generations with a burn-in of 25,000. The substitution model was selected on the basis of the corrected Akaike information criterion by ProtTest version 2.4 (http://darwin.uvigo.es/software/prottest.html).
Reverse transcription PCR for a 284-bp fragment in ORF2 of this HEV, which we named dromedary camel HEV (DcHEV), was positive for 3 fecal samples; viral loads were 3.7 × 105, 4.5 × 105, and 3.2 × 107 copies/mL. Complete-genome sequence data for 2 DcHEV strains (GenBank accession nos. KJ496143–KJ496144) revealed that the genome size was 7,220 bases and had a G+C content of 55% (Table). Overall, the DcHEV genomes differed from all other HEVs by >20% nt (Technical Appendix Table).
Figure 1
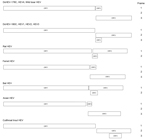
Figure 1. Predicted genomic organization of hepatitis E virus (HEV) from dromedary camel (DcHEV) and other HEVs, considering the reading frame of open reading frame (ORF) 1 as frame 1.
The DcHEV genome contained 3 major ORFs (Table, Figure 1). ORF1 polyprotein contained motifs consistent with a methyltransferase, a peptide containing a Y domain, a papain-like cysteine protease, a peptide with a hypervariable region (HVR), a helicase, and an RNA-dependent RNA polymerase. Also present in DcHEV were conserved sequences TLYTRTWS and RRLLXTYPDG, which bound the HVR of HEV1–4 and of 2 recently discovered wild boar HEV strains (10, 11) but not the HVR of ferret, rat, bat, avian or cutthroat trout HEVs. A conserved motif, (T/V)SGFSS(D/C)F(S/A)P, immediately preceding the HVR of only HEV3 and HEV4 was present in DcHEV as VSGFSSDFAP. The relative excess of proline and serine observed in the HVR of all other HEVs was also observed for DcHEV. For DcHEV strain 178C, ORF2 began at nt 5172, similar to what is found for HEV4 and wild boar HEV, with an insertion of a single nucleotide (U) at nt 5146, and ended at nt 7154, encoding a capsid protein of 660 aa (Technical Appendix Figure) (11–13). As for DcHEV strain 180C, because of the lack of the U insertion as in HEV1, HEV2, and HEV3, ORF2 began at nt 5171 (Technical Appendix Figure). For DcHEV strain 178C, similar to HEV4 and the 2 recently discovered wild boar HEV strains (11), ORF3 began at nt 5161 (Technical Appendix Figure) and ended at nt 5502, encoding a small phosphoprotein of 113 aa with a multifunctional C-terminal region. As for DcHEV strain 180C, because of the lack of the U insertion as in HEV1, HEV2, and HEV3, ORF3 began at nt 5160. The conserved cis-reactive element (UGAAUAACAUGU) located upstream of ORF2 and ORF3 in both strains might serve as promoter for the synthesis of the subgenomic mRNA for these 2 ORFs.
Figure 2

Figure 2. Phylogenetic analyses of open reading frame (ORF) 1 (A), ORF2 (B), ORF3 (C), and ORF1/ORF2 proteins, excluding the hypervariable region (HVR) (D) of hepatitis E virus (HEV) from dromedary camels (DcHEV)The...
Phylogenetic trees constructed by using ORF1, ORF2, ORF3, and concatenated ORF1/ORF2 excluding the HVR showed that DcHEV was clustered with different HEVs in different phylogenetic trees (Figure 2). For ORF1 and concatenated ORF1/ORF2 excluding the HVR, DcHEV was clustered with HEV3; but for ORF2 and ORF3, DcHEV was clustered with HEV1 and HEV2. Recombination analysis performed by using bootscan revealed no obvious and definite site of recombination, similar to what we observed in previous studies for other viruses (14), although different regions of the DcHEV genome might be more similar to different genotypes of HEV (data not shown).
We discovered HEV in dromedaries from the Middle East and named the virus DcHEV. In a recent study conducted in Dubai, HEV accounted for 40% of cases of acute hepatitis in humans (15). Although HEV is a major pathogen in the Middle East, sequence data for HEVs in the Arabian Peninsula are limited. The study reported here revealed that 1.5% of the adult dromedary fecal samples showed evidence of DcHEV RNA. Because humans come in close contact with dromedaries, our finding of DcHEV in dromedaries indicates a previously unknown potential reservoir and source of HEV infection for humans.
Comparative genomic and phylogenetic analyses showed that DcHEV probably represents a previously unrecognized HEV genotype. The conserved motif preceding the HVR in ORF1 resembled those found in HEV3, HEV4, and the 2 recently discovered wild boar HEV strains. Although phylogenetically ORF1 of DcHEV was clustered with HEV3, ORF2 and ORF3 of DcHEV were clustered with HEV1 and HEV2. Of note, ORF2 and ORF3 of the 2 DcHEV strains with complete genomes sequenced in this study resembled those of different HEV genotypes. The presence of a U insertion downstream to the second possible start codon for ORF2 (AUG2) in DcHEV strain 178C resembled the presence of a U insertion in HEV4 and wild boar HEV, leading to 3 possible start codons for its ORF2 but 1 possible start codon for its ORF3; whereas, the lack of this U insertion downstream to AUG2 in DcHEV strain 180C resembled the lack of U insertions in HEV1, HEV2 and HEV3, leading to only 1 possible start codon for its ORF2 but 3 possible start codons for its ORF3. To our knowledge, this presence or absence of such a U insertion in different strains of the same HEV has never been observed in other HEV genotypes and is unique to DcHEV. Although different regions of the DcHEV genome possessed characteristics associated with different kinds of HEV, no significant recombination was detected between DcHEV and the other HEVs. Because different regions of the genomes of DcHEV resembled those of different HEV genotypes, and even the genomes of different strains of DcHEV resembled those of different HEV genotypes, we propose that DcHEV should constitute a new HEV genotype.
Acknowledgments
We thank Wing-Man Ko and Constance Chan, for continuous support. We also thank Shanty Jose and Sweena Liddle for their help with sample preparation.
This work was partly supported by the Hong Kong Special Administrative Region Health and Medical Research Fund; Seed Funding for Theme-Based Research Scheme, and Strategic Research Theme Fund, The University of Hong Kong; Theme-based Research Scheme, Research Grant Council Grant, University Grant Council; and Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease for the Hong Kong Special Administrative Region Department of Health.
Dr Woo is professor and head of microbiology at The University of Hong Kong. His research focuses on novel microbe discovery and microbial genomics.
References
- Raj VS, Smits SL, Pas SD, Provacia LB, Moorman-Roest H, Osterhaus AD, Novel hepatitis E virus in ferrets, the Netherlands. Emerg Infect Dis. 2012;18:1369–70 . DOIPubMedGoogle Scholar
- Krog JS, Breum SO, Jensen TH, Larsen LE. Hepatitis E virus variant in farmed mink, Denmark. Emerg Infect Dis. 2013;19:2028–30. DOIPubMedGoogle Scholar
- Johne R, Heckel G, Plenge-Bonig A, Kindler E, Maresch C, Reetz J, Novel hepatitis E virus genotype in Norway rats, Germany. Emerg Infect Dis. 2010;16:1452–5. DOIPubMedGoogle Scholar
- Matsuda H, Okada K, Takahashi K, Mishiro S. Severe hepatitis E virus infection after ingestion of uncooked liver from a wild boar. J Infect Dis. 2003;188:944 . DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol. 2012;86:3995–4008. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Choi GK, Huang Y, Teng JL, Tsoi HW, Natural occurrence and characterization of two internal ribosome entry site elements in a novel virus, canine picodicistrovirus, in the picornavirus-like superfamily. J Virol. 2012;86:2797–808. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Wernery U, Wong EY, Tsang AK, Johnson B, Novel betacoronavirus in dromedaries of the Middle East, 2013. Emerg Infect Dis. 2014;20:560–72. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Lam CS, Tsang AK, Hui SW, Fan RY, Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in gammacoronavirus. J Virol. 2014;88:1318–31. DOIPubMedGoogle Scholar
- Smith DB, Purdy MA, Simmonds P. Genetic variability and the classification of hepatitis E virus. J Virol. 2013;87:4161–9. DOIPubMedGoogle Scholar
- Purdy MA, Lara J, Khudyakov YE. The hepatitis E virus polyproline region is involved in viral adaptation. PLoS ONE. 2012;7:e35974. DOIPubMedGoogle Scholar
- Takahashi M, Nishizawa T, Sato H, Sato Y. Jirintai, Nagashima S, et al. Analysis of the full-length genome of a hepatitis E virus isolate obtained from a wild boar in Japan that is classifiable into a novel genotype. J Gen Virol. 2011;92:902–8.
- Graff J, Torian U, Nguyen H, Emerson SU. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J Virol. 2006;80:5919–26. DOIPubMedGoogle Scholar
- Huang YW, Opriessnig T, Halbur PG, Meng XJ. Initiation at the third in-frame AUG codon of open reading frame 3 of the hepatitis E virus is essential for viral infectivity in vivo. J Virol. 2007;81:3018–26. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol. 2006;80:7136–45. DOIPubMedGoogle Scholar
- Abro AH, Abdou AM, Saleh AA, Ustadi AM, Hussaini HS. Hepatitis E: a common cause of acute viral hepatitis. J Pak Med Assoc. 2009;59:92–4 .PubMedGoogle Scholar
Figures
Table
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 20, Number 6—June 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Patrick C.Y. Woo, State Key Laboratory of Emerging Infectious Diseases, Department of Microbiology, The University of Hong Kong, University Pathology Building, Queen Mary Hospital, Hong Kong, China
Top