Volume 21, Number 2—February 2015
Research
Microbiota That Affect Risk for Shigellosis in Children in Low-Income Countries
Cite This Article
Citation for Media
Abstract
Pathogens in the gastrointestinal tract exist within a vast population of microbes. We examined associations between pathogens and composition of gut microbiota as they relate to Shigella spp./enteroinvasive Escherichia coli infection. We analyzed 3,035 stool specimens (1,735 nondiarrheal and 1,300 moderate-to-severe diarrheal) from the Global Enteric Multicenter Study for 9 enteropathogens. Diarrheal specimens had a higher number of enteropathogens (diarrheal mean 1.4, nondiarrheal mean 0.95; p<0.0001). Rotavirus showed a negative association with Shigella spp. in cases of diarrhea (odds ratio 0.31, 95% CI 0.17–0.55) and had a large combined effect on moderate-to-severe diarrhea (odds ratio 29, 95% CI 3.8–220). In 4 Lactobacillus taxa identified by 16S rRNA gene sequencing, the association between pathogen and disease was decreased, which is consistent with the possibility that Lactobacillus spp. are protective against Shigella spp.–induced diarrhea. Bacterial diversity of gut microbiota was associated with diarrhea status, not high levels of the Shigella spp. ipaH gene.
Diarrheal disease contributes substantially to illness and death in children in low-income countries (1,2). Recent investigations of enteric illness have shown many cases with >1 pathogen identified (3–5). The paradigm of 1 pathogen and 1 disease has been questioned with the advent of microbiological and molecular detection methods that have lower limits of detection. Children in developing countries are exposed to an array of pathogenic organisms. Recent studies have shown a complex relationship between gut microbiota and diarrheal illness; children with severe illness tend to have a less diverse microbiota and a predominance of specific genera of organisms (6). Molecular-based approaches to pathogen detection enhance the ability to quantify the abundance of pathogens shed in the stool.
Two recent studies of children in low-income countries have highlighted the need for pathogen quantitation. Lindsay et al., using the Global Enteric Multicenter Study (GEMS) specimen collection, found that 80% of controls and 89% of case-patients had detectable levels of Shigella spp. (7). To identify which children had shigellosis, Lindsay et al. determined a quantitative threshold and, when applied, it identified twice as many cases compared with standard culture. Platts-Mills et al., in a study of populations with a high prevalence of malnutrition and enteric infections in Tanzania, compared samples taken before and during diarrheal episodes (8). They did not find an association between the presence of any pathogen and diarrhea for 15 pathogens studied (rotavirus, adenovirus, astrovirus, norovirus, sapovirus, Cryptosporidium spp., Giardia lamblia, Campylobacter jejuni, Clostridium difficile, Salmonella spp., Shiga-toxigenic Escherichia coli, Shigella spp./enteroinvasive E. coli [EIEC], enterotoxigenic E. coli [ETEC], typical enteropathogenic E. coli [tEPEC], and enteroaggregative E. coli [EAEC]). However, when they considered quantity of pathogen on a continuous scale, 3 organisms (rotavirus, astrovirus, and Shigella spp.) were associated with diarrhea.
In disease-endemic settings, detection of multiple enteropathogens in asymptomatic and symptomatic children is common (4,9). Samples from one third of patients with diarrhea in a hospital study in Kolkata, India, contained >1 pathogen. Negative associations were demonstrated between Shigella spp. and rotavirus and Shigella spp. and Vibrio cholerae (3). However, a limitation of this study was that it was conducted only in patients with diarrhea. Thus, differential comparisons could not be made between pathogen associations in diarrheal and nondiarrheal samples. A recent study by Taniuchi et al. reported the etiology of diarrheal episodes by using molecular methods in Bangladeshi children during their first year of life. They found that multiple enteropathogens were present by the first month of life in stool specimens from healthy children and from children with diarrhea (4). If multiple pathogens are present, they might interact to increase or decrease the probability of symptomatic infection. Bhavnani et al. reported synergistic effects in rotavirus–G. lamblia and rotavirus–E. coli infections, in which the presence of these co-occurring organisms increased the probability of disease (10).
The gut microbiota are composed of thousands of species that might play a role in the risk for diarrhea. Some Lactobacillus and Veillonella species are potentially protective against diarrhea or serve as markers of healthy gut microbiota (11–13) Probiotic activity has been associated with some Lactobacillus spp., bifidobacteria, Veillonella spp., Streptococcus spp., Enterococcus spp., nonpathogenic E. coli, and Saccharomyces boulardii (13). Randomized clinical trials have investigated the role of some Lactobacillus spp. in treating infectious diarrhea and identified that these organisms can provide a benefit in the treatment of acute, infectious, watery diarrhea in infants and young children (12). In this study, we examined relationships between Shigella spp./EIEC, microbiota, and diarrhea by using 16S rRNA marker gene surveys of stool specimens from a large international study of diarrhea in children <5 years of age (9).
Study Design and Participants
We used stool specimens collected from children <5 years of age who participated in a matched case–control study of moderate-to-severe diarrhea sponsored by the GEMS consortium (14,15). In brief, the GEMS was a prospective case–control study of infants and young children at 7 sites in sub-Saharan Africa and southern Asia. Case-patients with moderate-to-severe diarrhea were enrolled when they came to a health clinic. Moderate-to-severe diarrhea eligibility criteria included dehydration (sunken eyes, loss of normal skin turgor, or a decision to initiate intravenous hydration), the presence of blood in the stool (dysentery), or a clinical decision to hospitalize the child. In the GEMS, matching controls (for sex, age, and community) were sampled from a demographic surveillance database of the area and included if they reported no diarrhea within the previous 7 days. For this study, 4 of the 7 GEMS sites (The Gambia, Mali, Kenya, and Bangladesh) elected to participate in further molecular characterization of their samples. When samples were sent for analysis, it was not required that their matched specimen be included, which resulted in disruption of matches. Including only samples with the complete matched set would have limited our sample size. Therefore, we included age and location as covariates in our analysis but did not analyze samples as matched sets.
All specimens for this study were collected during December 2007–December 2009. One specimen was collected for each child at the time of enrollment. The Institutional Review Boards at all participating institutions reviewed and approved the protocol.
Specimen Collection
Stool specimens were handled according to the GEMS protocol (16). DNA was isolated from frozen stool specimens by using a bead beater with 3-mm diameter solid glass beads (Sigma-Aldrich, St. Louis, MO, USA) and, subsequently, with 0.1-mm zirconium beads (BioSpec Products, Inc., Bartlesville, OK, USA) to disrupt cells. The cell slurry was then centrifuged at 16,000 × g for 1 min, and the supernatant was processed by using QIAamp DNA Stool Extraction Kit (QIAGEN, Valencia, CA, USA). Extracted DNA was precipitated with ethanol and shipped to the United States.
Detection of EAEC, tEPEC, ETEC, Rotavirus, Norovirus, G. lamblia, and Cryptosporidium spp.
Diagnostic microbiological methods for rotavirus, norovirus, G. lamblia, Cryptosporidium spp., and diarrheagenic E. coli (EAEC, tEPEC, and ETEC) were conducted at each site as described by Panchalingham et al. (16). In brief, E. coli isolates were selected from MacConkey agar plates and tested by using motility indole ornithine medium. EAEC, tEPEC, and ETEC pathotypes were identified by using PCRs for known virulence determinants of each pathogen (ETEC: heat-labile and heat-stable enterotoxins; tEPEC: intimin [eae] and bundle-forming pilus; and EAEC: aatA and aaiC genes).
Rotavirus was detected by using the ProSpecT ELISA Rotavirus Kit (Oxoid, Basingstoke, UK). Norovirus genogroups GI and GII were detected by using a multiplex PCR specific for synthesized complementary DNA after viral RNA extraction and reverse transcription. G. lamblia and Cryptosporidium spp. were detected by using commercially available immunoassays (TechLab, Blacksburg, VA, USA) following the manufacturer’s protocols. Although the GEMS study tested for other enteric pathogens, our analysis included only pathogens that were present in ≥10 specimens. This lower limit was chosen after examination of the distribution of positive samples for each pathogen and to avoid statistical testing with small sample sizes.
Quantitative PCR for Detection of ipaH Gene in Shigella spp. and EIEC
Each stool DNA specimen was tested by using a quantitative PCR (qPCR) for Shigella/EIEC that included the 7500/700 Fast Real-Time PCR System, software V2·0·5, and SYBR green–based fluorescent dye (Applied Biosystems, Foster City, CA, USA). Details on primer design, PCR conditions, and standard curve analysis have been reported elsewhere (4,7,17). Shigella/EIEC was identified by using primers specific for the ipaH gene (7). We used a cutoff of 14,000 ipaH gene copies to distinguish children shedding low levels of Shigella spp. from those shedding high levels of Shigella spp. in their stools. The threshold was established by constructing receiver-operating characteristic curves to determine sensitivity and specificity of incremental increases in levels of ipaH compared with disease status and set on the basis of the point that maximized sensitivity and specificity (7). Stool specimens with high and low levels of ipaH indicate the relative amount of Shigella spp. detected.
16S rRNA Gene Sequencing and Analysis and Identification of C. jejuni
DNA was amplified by using universal primers specific for the V1–V3 region of the 16S rRNA gene in bacteria (518R [5′-CAATTACCGCGGCTGCTGG-3′] and 27F [5′-AGAGTTTGATCCTGGCTCAG-3′]). Individual reads were filtered for quality by using custom in-house scripts that removed low-quality sequences as described (6). Remaining high-quality sequences were separated into sample-specific sets according to barcodes. Conservative operational taxonomic units (OTUs) were clustered by using DNACLUST with parameters (–r 1 and 99% identity clusters) to ensure that the definition of an OTU was consistent across all samples (18). For taxonomic identification, a representative sequence from each OTU was aligned to the Ribosomal Database (RDP) (rdp.cme.msu.edu, release 10.4) by using BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with long word length (–W 100) to detect only nearly identical sequences (19). Sequences without a nearly identical match to the RDP (>100-bp perfect match and >97% identity, as defined by BLAST) were marked as being unassigned and assigned a unique OTU identifier. If a sample contained taxa classified as C. jejuni, we identified this sample as positive for C. jejuni.
Statistical Analysis
Associations between high levels of ipaH and each additional pathogen, stratified by diarrheal status, were assessed for children with moderate or severe diarrhea and for controls by using separate logistic regression models with high levels of ipaH as the dependent variable. Logistic regression with moderate-to-severe diarrhea as the outcome of interest was then conducted to test for the interaction between high levels of ipaH and either 1) pathogenic microorganisms tested for by the GEMS or 2) species identified by 16S rRNA gene sequencing. All models were adjusted for potential confounding caused by location and age with categorical location and age terms in the model.
Statistical modeling was used to examine whether microbes interact to effect diarrhea risk. To assess whether the risk caused by having Shigella spp. and an additional microbe differed from the product of the risks caused by having each microbe separately (i.e., multiplicative interaction), we used a logistic regression model with an interaction term containing the level of ipaH and the additional microbe of interest (Technical Appendix Figure). To assess whether the excess risk of having ipaH and an additional microbe differed from the sum of the excess risk of having each separately (i.e., additive interaction), we estimated the relative excess risk caused by the interaction (RERI, also called the interaction contrast ratio [ICC]) (20–22). An RERI = 0 indicates no additive interaction, an RERI>0 suggests positive additive interaction, and an RERI<0 suggests negative additive interaction. Ninety-five percent CIs were estimated by using the Hosmer-Lemeshow procedure (23,24). All statistical analysis was performed by using SAS version 9·2 (SAS Institute, Cary, NC, USA) and R 2.15 (25), and p values <0.05 were considered significant.
A total of 3,035 (1,735 nondiarrheal and 1,300 diarrheal) stool specimens from children <5 years of age from The Gambia, Mali, Kenya, and Bangladesh were examined for Shigella spp. by identification of ipaH by qPCR; for G. lamblia, Cryptosporidium spp., and rotavirus by ELISA; for norovirus by PCR; for EAEC, ETEC, or tEPEC by culture and subsequent PCR; and for C. jejuni by 16S rRNA gene sequencing. Characteristics of samples are shown in Table 1. Diarrheal samples had a significantly higher number of pathogens (diarrheal mean 1.4, nondiarrheal mean = 0.95; p<0.05).
Associations and Interaction Effects of Co-occurring Pathogens
Figure 1

Figure 1. Association of co-occurring pathogens with high levels of ipaH gene of Shigella spp. in stool specimens of children with diarrhea (cases) and children without diarrhea (controls) in low-income countries. Dark circles...
In 69% (278/404) of samples with high levels of ipaH (71% of diarrheal samples with high levels of ipaH and 64% nondiarrheal samples with high levels of ipaH), we identified an additional pathogen. In samples with low levels of ipaH, we identified an additional pathogen in 69% (1,815/2,631; 80% of diarrheal samples with low levels of ipaH and 62% of nondiarrheal samples with low levels of ipaH). After adjusting for age and location, rotavirus exhibited a negative association with high levels of ipaH in case-patients (odds ratio [OR] 0.31, 95% CI 0.17–0.55). In case-patients and controls, no other pathogen showed an association (Figure 1; Technical Appendix Table 1).
Of the 8 pathogens tested, none showed any interaction effects. However, rotavirus was negatively associated with high levels of ipaH (Table 2), and only 15 samples that had high levels of ipaH were positive for rotavirus; 14 of these 15 samples were diarrheal samples. The presence of rotavirus and high levels of ipaH resulted in an OR of 29 (95% CI 3.8–220) for diarrheal risk. Although this point estimate was much higher than the expected additive effect of 11, this result and results of tests for interaction were not statistically significant (Table 3).
Interaction Effects of Lactobacillus and Veillonella Taxa
Figure 2
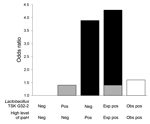
Figure 2. Departure from additivity between level of ipaH gene and presence of Lactobacillus taxon TSK G32-2 on odds of moderate-to-severe diarrhea in children in low-income countries. The reference group is TSK G32-2...
A total of 61 Lactobacillus and Veillonella taxa were identified by 16S rRNA gene sequencing. Analyses were limited to 31 taxa that co-occurred in samples with high levels of ipaH ≥10 times. We tested for interaction between high levels of ipaH and the identified 16S rRNA gene taxon by using a logistic regression model adjusted for age and location, including an interaction term between level of ipaH and taxon presence (Technical Appendix Table 2). Five taxa showed significant negative multiplicative interactions identified by an interaction term with p ≤ 0·05. Of these taxa, Lactobacillus ruminis (RERI −1.92, 95% CI −3.36 to 0.47), Lactobacillus DJF-RP24 (RERI −2.44, [95% CI −3.93 to 0.95), Lactobacillus KLDS 1.0718 (RERI −1.93, 95% CI −3.56 to 0.29), and Lactobacillus TSK G32.2 (RERI −2.69, 95% CI −4.55 to 0.84]) showed additive interactions (Table 3). The combined effect of high levels of ipaH in the presence of L. ruminis, Lactobacillus DJF RP24, Lactobacillus KLDS 1.0718, or Lactobacillus TSK G32.2 was lower than expected (Figure 2; Technical Appendix Table 3), which suggested an antagonistic interaction or a decreased association with diarrhea when specific Lactobacillus taxa were present than versus when they were absent. When we tested the 4 Lactobacillus taxa against 8 additional pathogens; no additive interaction was observed (Technical Appendix Table 4).
16S rRNA Gene–based Bacterial Community Profiles
Figure 3

Figure 3. Overall 16S rRNA gene–based bacterial community profiles (proportional abundance) of diarrheal samples with high levels of ipaH gene (n = 277), diarrheal samples with low levels of ipaH gene (n =...
The proportional abundance of the 9 most common genera identified by 16S rRNA marker gene sequencing differed among stool specimens with high and low levels of ipaH and among specimens from children with and without diarrhea (Figure 3). Overall composition of diarrheal stool specimens had an increased relative proportional abundance of facultative anaerobes when compared with composition of nondiarrheal stool specimens, even when diarrheal stool specimens had low levels of ipaH (mean diarrheal specimens 0.47, mean nondiarrheal specimens 0.22; p<0.0001). Overall, diarrheal stool specimens with high levels of ipaH had the lowest proportion abundance of Prevotella spp. (0.11), diarrheal stool samples with low levels of ipaH had the second lowest proportional abundance (0.15), and nondiarrheal stool specimens with high and low levels of ipaH had similar proportion abundances for Prevotella spp. (0.25 and 0.24, respectively; p = 0.63).
Shigella spp. identified by the ipaH qPCR were a subset of the genera Escherichia/Shigella identified by 16S rRNA gene sequencing, but could not be distinguished from commensal strains. Diarrheal specimens had a larger proportion of members of the genera Escherichia/Shigella (p<0.0001) than nondiarrheal specimens, regardless of levels of ipaH. However, diarrheal stool specimens with high levels of ipaH had a significantly higher proportion of Escherichia/Shigella sequences (31%; p<0.0001) than did diarrheal stool specimens with low levels of ipaH.
Figure 4
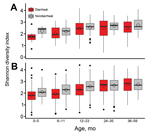
Figure 4. Shannon diversity index for diarrheal and nondiarrheal samples with high (A) and low (B) levels of Shigella spp. ipaH gene stratified by age group for children in low-income countries. Box...
Members of the genus Veillonella was found equally in diarrheal and nondiarrheal stool specimens. Streptococci were found more often in diarrheal stool specimens, regardless of levels of ipaH. There was no association between Shannon diversity indices and levels of ipaH (p = 0.95), although diversity was significantly associated with age, location, and moderate-to-severe diarrhea (p<0.0001, p = 0.004, and p<0.0001, respectively) (Figure 4).
In this study, we used qPCR and 16S rRNA gene sequencing to identify interactions between Shigella/EIEC and co-occurring enteric pathogens or microbes within the gut microbiota in children with moderate-to-severe diarrhea. We explored these interactions in stool specimens obtained from children with and without diarrhea who participated in the international GEMS study. We used detection of the ipaH gene as an indicator of Shigella/EIEC infection because this molecular method is highly sensitive and specific (4,26,27). Use of quantitative identification methods, rather than colonization (7,8), is advantageous for identifying true disease associations. We found that 69% (1,815/2,631) of stool samples with high levels of ipaH had a co-occurring pathogen.
This study confirms the findings of the negative association between rotavirus and Shigella spp. in polymicrobial infections in diarrheal patients in India and The Gambia (Shigella spp./rotavirus: OR 0.36, 95% CI 0.14–0.92) (3,28). Although we identified correlations between rotavirus and Shigella spp. as determined by levels of ipaH, no pathogen showed an antagonistic or synergistic interaction on the odds of moderate-to-severe diarrheal illness. Although all but 1 of the rotavirus-positive samples that had high levels of ipaH were associated with diarrhea cases, the negative association between these pathogens gave us limited sample size to assess a possible synergistic effect. A synergistic effect between high levels of ipaH and rotavirus (RERI/ICC 9.9, 95% CI 2.6–28.4) was previously observed in Ecuador, and it was concluded that pathogenic potential appears to be enhanced during co-infection (10). Our RERI/ICC was 18, but the effect was not significant, possibly because of small sample size.
The GEMS was designed as a matched case–control study of thousands of stool samples from persons matched by age, sex, community, and time. A possible limitation of our methods is that we did not use matched pairs but rather adjusted for country-level location and age by using statistical modeling. This method was used because of the large number of broken matched pairs. Thus, inclusion of all samples greatly increased our sample size. Generally, the lack of an appropriate matched analysis biases the effect on estimates toward the null. However, studies have shown that including samples with missing data and adjusting by using confounding terms is valuable, particularly with a larger sample size (29,30). We adjusted for location at the country level, and the GEMS matched case-patients at the community level, which could fail to adequately address bias, particularly when one considers that the distribution of case-patients and controls differed at multiple sites and that Shigella spp. are more common in Bangladesh. An additional limitation of our study design is that, given a cross-sectional study sample set, we were unable to identify temporal associations and to attribute cause and effect. Furthermore, associations between pathogens and taxa may be explained by seasonality, and further work should be conducted to investigate this as a possible explanation. Finally, the 16S rRNA gene-sequencing method used to identify taxa of interest is limited because our identification was only as precise as the RDP attributed taxonomy. Thus, further characterization and species-specific identification are warranted in uncultured bacteria such as Lactobacillus KLDS 1.0718.
A previous study showed cross-sectional differences in stool microbiota of children with diarrhea compared with children without diarrhea in low-income countries (6). Our study showed that the composition of microbiota is more closely associated with diarrheal status than with co-infection by Shigella spp./EIEC as measured by high levels of ipaH. Microbiota in stools of children without diarrhea but who had high levels of ipaH (i.e., were colonized with Shigella spp./EIEC) were more similar to microbiota of healthy children with low levels of ipaH than microbiota in samples from children with diarrhea and high levels of ipaH.
In low-income countries, infection/colonization with pathogens occurs commonly in persons without diarrhea. Our study found little evidence of interaction between Shigella spp. and co-occurring pathogens. Although the cross-sectional study design precludes strong statements of cause and effect, our data are consistent with the possibility that some Lactobacillus taxa naturally occurring in the gut and are protective against Shigella spp.–induced diarrhea. Future studies should continue to consider the effects of co-occurring species.
This study was supported by the Bill and Melinda Gates Foundation (awards BMGF 38874 to M.M.L., K.K., and J.P.N.; BMGF OPP42917 to J.P.N. and O.C.S.; and BMGF OPP1016839 to J.P.N. and O.C.S.); the National Institutes of Health (grant U19 090873 to James Kaper); and the US National Science Foundation (Graduate Research Fellowship DGE0750616 to J.N.P.).
At the time of this study, Dr. Lindsay was a research analyst and doctoral student at the University of Maryland, Baltimore, Maryland. Her research interests are identification and quantitation of Shigella spp. and how this pathogen interacts with human gut microbiota.
Acknowledgments
We thank Carol Tackett for her consultation on clinical terminology and readability.
B.L., J.P.N., and O.C.S. developed the hypotheses; M.M.L., K.K., and J.P.N. led the Global Enteric Multicenter Study; B.L., J.O., M.A.H., M.A., B.T., R.O., A.S.G.F., S.K.D., U.N.I., M.A., D. Sanogo, D. Saha, S.S., T.H.F., D.N., and S.P. collected data and developed and maintain databases; A.W.W., J.N.P., J.P., M.P., and O.C.S. developed and performed 16S rRNA gene sequencing, assembly, and taxonomic identification; S.L., B.L., and S.P. performed laboratory protocols; and B.L., L.S.M., L.H., and H.S. performed analyses.
References
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–128. DOIPubMedGoogle Scholar
- Walker CL, Aryee MJ, Boschi-Pinto C, Black RE. Estimating diarrhea mortality among young children in low and middle income countries. PLoS ONE. 2012;7:e29151. DOIPubMedGoogle Scholar
- Lindsay B, Ramamurthy T, Sen Gupta S, Takeda Y, Rajendran K, Nair GB, Diarrheagenic pathogens in polymicrobial infections. Emerg Infect Dis. 2011;17:606–11. DOIPubMedGoogle Scholar
- Taniuchi M, Sobuz SU, Begum S, Platts-Mills JA, Liu J, Yang Z, Etiology of diarrhea in Bangladeshi infants in the first year of life analyzed using molecular methods. J Infect Dis. 2013;208:1794–802. DOIPubMedGoogle Scholar
- Platts-Mills JA, Liu J, Houpt ER. New concepts in diagnostics for infectious diarrhea. Mucosal Immunol. 2013;6:876–85. DOIPubMedGoogle Scholar
- Pop M, Walker AW, Paulson JN, Lindsay B, Antonio M, Hossain MA, Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiome composition. Genome Biol. 2014;15:R76. DOIPubMedGoogle Scholar
- Lindsay B, Ochieng JB, Ikumapayi UN, Toure A, Ahmed D, Li S, Quantitative PCR for detection of Shigella improves ascertainment of Shigella burden in children with moderate-to-severe diarrhea in low-income countries. J Clin Microbiol. 2013;51:1740–6. DOIPubMedGoogle Scholar
- Platts-Mills JA, Gratz J, Mduma E, Svensen E, Amour C, Liu J, Association between stool enteropathogen quantity and disease in Tanzanian children using TaqMan array cards: a nested case-control study. Am J Trop Med Hyg. 2014;90:133–8. DOIPubMedGoogle Scholar
- Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–22. DOIPubMedGoogle Scholar
- Bhavnani D, Goldstick JE, Cevallos W, Trueba G, Eisenberg JNS. Synergistic effects between rotavirus and coinfecting pathogens on diarrheal disease: evidence from a community-based study in northwestern Ecuador. Am J Epidemiol. 2012;176:387–95. DOIPubMedGoogle Scholar
- Yun J-H, Yim D-S, Kang J-Y, Kang B-Y, Shin E-A, Chung M-J, Identification of Lactobacillus ruminus SPM0211 isolated from healthy Koreans and its antimicrobial activity against some pathogens. Arch Pharm Res. 2005;28:660–6. DOIPubMedGoogle Scholar
- Guandalini S. Probiotics for children with diarrhea: an update. J Clin Gastroenterol. 2008;42(Suppl 2):S53–7. DOIPubMedGoogle Scholar
- Srikanth CV, McCormick BA. Interactions of the intestinal epithelium with the pathogen and the indigenous microbiota: a three-way crosswalk. Interdiscip Perspect Infect Dis. 2008;2008:626827. . Epub 2008 Oct 29.DOIGoogle Scholar
- Levine MM, Kotloff KL, Nataro JP, Muhsen K. The Global Enteric Multicenter Study (GEMS): impetus, rationale, and genesis. Clin Infect Dis. 2012;55(Suppl 4):S215–24. DOIPubMedGoogle Scholar
- Kotloff KL, Blackwelder WC, Nasrin D, Nataro JP, Farag TH, van Eijk A, The Global Enteric Multicenter Study (GEMS) of diarrheal disease in infants and young children in developing countries: epidemiologic and clinical methods of the case/control study. Clin Infect Dis. 2012;55(Suppl 4):S232–45. DOIPubMedGoogle Scholar
- Panchalingam S, Antonio M, Hossain A, Mandomando I, Ochieng B, Oundo J, Diagnostic microbiologic methods in the GEMS-1 case/control study. Clin Infect Dis. 2012;55(Suppl 4):S294–302. DOIPubMedGoogle Scholar
- Vu DT, Sethabutr O, Von Seidlein L, Tran VT, Do GC, Bui TC, Detection of Shigella by a PCR assay targeting the ipaH gene suggests increased prevalence of shigellosis in Nha Trang, Vietnam. J Clin Microbiol. 2004;42:2031–5. DOIPubMedGoogle Scholar
- Ghodsi M, Liu B, Pop M. DNACLUST: accurate and efficient clustering of phylogenetic marker genes. BMC Bioinformatics. 2011;12:271 and. DOIPubMedGoogle Scholar
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 2005;33:D294–6. DOIPubMedGoogle Scholar
- Rothman KJ. Epidemiology: an introduction. New York: Oxford University Press; 2012.
- Knol MJ, VanderWeele TJ. Recommendations for presenting analyses of effect modification and interaction. Int J Epidemiol. 2012;41:514–20. DOIPubMedGoogle Scholar
- Andersson T, Alfredsson L, Källberg H, Zdravkovic S, Ahlbom A. Calculating measures of biological interaction. Eur J Epidemiol. 2005;20:575–9. DOIPubMedGoogle Scholar
- Zou GY. On the estimation of additive interaction by use of the four-by-two table and beyond. Am J Epidemiol. 2008;168:212–24. DOIPubMedGoogle Scholar
- Hosmer DW, Lemeshow S. Confidence interval estimation of interaction. Epidemiology. 1992;3:452–6 and. DOIPubMedGoogle Scholar
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Vegan: Community Ecology Package, 2013 [cited 2014 Apr 8]. http://cran.r-project.org/web/packages/vegan/index.html
- Lindsay B, Pop M, Antonio M, Walker AW, Mai V, Ahmed D, Survey of culture, goldengate assay, universal biosensor assay, and 16S rRNA gene sequencing as alternative methods of bacterial pathogen detection. J Clin Microbiol. 2013;51:3263–9. DOIPubMedGoogle Scholar
- Sinha A, SenGupta S, Guin S, Dutta S, Ghosh S, Mukherjee P, Culture-independent real-time PCR reveals extensive polymicrobial infections in hospitalized diarrhoea cases in Kolkata, India. Clin Microbiol Infect. 2013;19:173–80. DOIPubMedGoogle Scholar
- Kwambana BA, Ikumapayi UN, Sallah N, Dione M, Jarju S, Panchalingham S, High genotypic diversity among rotavirus strains infecting Gambian children. Pediatr Infect Dis J. 2014;33(Suppl 1):S69–75. DOIPubMedGoogle Scholar
- Lynn HS, McCulloch CE. When does it pay to break the matches for analysis of a matched-pairs design? Biometrics. 1992;48:397–409. DOIPubMedGoogle Scholar
- Hansson L, Khamis HJ. Matched samples logistic regression in case-control studies with missing values: when to break the matches. Stat Methods Med Res. 2008;17:595–607 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 21, Number 2—February 2015
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Brianna Lindsay, Department of Epidemiology and Preventive Medicine, University of Maryland School of Medicine, 660 W Redwood St, Howard Hall, Rm 585, Baltimore, MD 21201, USA
Top