Volume 23, Number 3—March 2017
Research
Whole-Genome Analysis of Bartonella ancashensis, a Novel Pathogen Causing Verruga Peruana, Rural Ancash Region, Peru
Cite This Article
Citation for Media
Abstract
The genus Bartonella contains >40 species, and an increasing number of these Bartonella species are being implicated in human disease. One such pathogen is Bartonella ancashensis, which was isolated in blood samples from 2 patients living in Caraz, Peru, during a clinical trial of treatment for bartonellosis. Three B. ancashensis strains were analyzed by using whole-genome restriction mapping and high-throughput pyrosequencing. Genome-wide comparative analysis of Bartonella species showed that B. ancashensis has features seen in modern and ancient lineages of Bartonella species and is more related to B. bacilliformis. The divergence between B. ancashensis and B. bacilliformis is much greater than what is seen between known Bartonella genetic lineages. In addition, B. ancashensis contains type IV secretion system proteins, which are not present in B. bacilliformis. Whole-genome analysis indicates that B. ancashensis might represent a distinct Bartonella lineage phylogenetically related to B. bacilliformis.
Bartonelloses are major emerging infectious bacterial diseases because of the high prevalence of chronic Bartonella infections in mammals and humans and their increasing risk for infection of immunocompromised populations (1–4). Bartonella species are present worldwide and are associated with several diseases, such as Carrion’s disease, caused by B. bacilliformis; cat-scratch disease, caused by B. henselae; and trench fever, caused by B. quintana (2,5–7). B. henselae and B. quintana are also associated with chronic infections, such as bacillary angimatosis and endocarditis, and B. quintana chronic bacteremia is found in homeless populations worldwide (3,8–12).
In addition to these 3 major infectious pathogens, an increasing number of new Bartonella spp. have been identified in recent years as zoonotic pathogens transmitted by diverse arthropod vectors (1,2,7). Currently, >40 official and candidate Bartonella species are listed in the Taxonomy Database of the National Center for Biotechnology Information (http://www.bacterio.net/Bartonella.html); 31 are recognized species. During their evolution, Bartonella lineages have adapted to a variety of hosts and developed virulence factors associated with a diverse set of disease signs and symptoms (13–15).
Despite the high mortality rate for bartonellosis in Peru, studies on Bartonella pathogens are insufficient and mainly focused on B. bacilliformis (16). We previously reported identification of non-bacilliformis Bartonella in Peru during a clinical treatment trial (17–19). Using single-locus sequence typing, we identified 3 isolates (designated 20.00, 20.60, and 41.60) from 4 whole blood specimens collected from 2 patients (nos. 20 and 41) at day 0 or 60 during the clinical treatment trial in the Ancash region of Peru and found that these isolates had citrate synthase (gltA) gene sequences that diverged from that of B. bacilliformis reference strain KC583 (17–19). Multilocus sequencing typing and microbiological analyses indicated these 3 isolates are members of a novel Bartonella species, subsequently name B. ancashensis (17).
In this study, we performed genomic analyses of 3 non-bacilliformis Bartonella isolates obtained from patient blood samples (isolates 20.00, 20.60, and 41.60). Whole-genome analyses confirmed our previous identification of the isolates as a new species (B. ancashensis) (20) and identified unique genomic characteristics of B. ancashensis and differences between B. ancashensis and its closest relative (B. bacilliformis).
Ethics Statement
The human subject use protocol, including clinical specimen collection, and the consent procedure were approved in 2002 by the Institutional Review Boards of the Uniformed Services University of the Health Sciences (Bethesda, MD, USA); the Naval Medical Research Center Institutional Review Board (Bethesda, MD, USA); and the Universidad Peruana Cayetano Heredia (Lima, Peru). The trial details are available in the International Standard Randomized Controlled Trial Number registry (https://www.isrctn.com; trial no. ISRCTN16597283). Patients enrolled were 1–60 years of age, and written consent was obtained from the patient or the parent or guardian of the patient enrolled in the study.
Study Protocol
A clinical trial to compare rifampin, the standard drug for treatment of bartonellosis caused by chronic B. bacilliformis infection, with azithromycin, a possible alternative drug, was conducted in 2003 in the Caraz District of the Ancash region of Peru (Blazes DL, trial no. ISRCTN16597283). Patients with suspected chronic B. bacilliformis infection (verruga peruana) either came to the local hospital in Caraz or were identified by home visits. Patients (>1 year of age) with verruga peruana were randomly chosen to receive either a daily dose of rifampin (Pfizer, New York, NY, USA) for 14 days or 2 weekly doses of azithromycin (Pfizer) on days 0 and 7. For the patients who participated in the trial, survey data and medical records were collected on day 0 (baseline; time of presentation at the local hospital), and patients were then given the 2-week antimicrobial drug treatment. Clinical data was also collected on days 7, 14, 30, and 60.
In addition, peripheral blood specimens were collected from each patient on days 0, 7, 14, and 60 into tubes containing sodium citrate solution at the local hospital in Caraz and transported on ice to the clinical laboratory at the US Naval Medical Research Unit No. 6 (Lima, Peru) for blood cultures and analysis by PCR. Selected specimens, Bartonella isolates, and genomic DNA extracts were sent to the Naval Medical Research Center (Silver Spring, MD, USA) and the Walter Reed Army Institute of Research (Silver Spring, MD, USA) for additional investigations.
Blood specimens were cultured for >8 weeks as described (17–19). Bartonella culture-positive specimens were confirmed to be Bartontella species by using microbiological observations and molecular assays. Nucleic acids were isolated from culture-positive blood samples and subjected to PCR amplification of a 338-bp fragment of the gltA gene. The PCR product was sequenced by using the Sanger method.
We aligned partial gltA gene sequences (homologous to nt 781–1137 of the B. bacilliformis KC583 gltA gene) and used them for phylogenetic analyses. Samples with gltA sequences that showed major differences (>85% divergence) from those of B. bacilliformis were cultured on brain heart infusion agar supplemented with 10% defibrinated sheep blood (BD Diagnostics, Glencoe, MD, USA) for 10–28 days at the Naval Medical Research Center.
For next-generation sequencing (NGS), genomic DNA was extracted from Bartonella isolates, randomly fragmented by using focused ultrasonication (S2 System; Covaris, Inc., Woburn, MA, USA), and used in rapid shotgun genomic DNA library preparation and pyrosequencing with the 454 GS FLX Titanium System (Roche 454 Life Sciences, Branford, CT, USA). For whole-genome restriction map (WGRM) analysis, Bartonella isolates were freshly grown on brain heart infusion agar with 10% sheep blood at 30°C in an atmosphere of 5% CO2 for 10–14 days. High molecular weight DNA was isolated by using the Argus Sample Preparation Kit (OpGen Inc., Gaithersburg, MD, USA). DNA quality and quantity were assessed by using the Argus QCard Kit and mapped by using the Argus MapCard Kit and Argus Enzyme Kit-AflII with the Argus System (OpGen Inc.). Downstream analysis, clustering, and genome alignment was performed by using MapSolver version 3.2.4. (OpGen Inc.).
We de novo assembled NGS data into sequence contigs by using GS Assembler software version 2.5.3 (Newbler; https://wikis.utexas.edu/display/bioiteam/GS+De+novo+assembler) and then assembled the contigs to scaffolds with the WGRM as the physical reference. PCR amplification and open reading frame annotation were used to complete genome assembling. After we virtually digested the complete genome sequence with AflII, we aligned the in silico whole-genome restriction map with the WGRM to ensure the correct order and orientation of the final assemblies (21).
We performed pairwise genome-wide comparative analysis by comparing homologous proteins from pairs of Bartonella species. Results are presented as density distribution curves for amino acid identity and as dot plots for pairwise amino acid identity for each homologous protein. Genes of interest were subjected to additional analysis, including gene cluster comparisons. For genome-wide phylogenetic analysis of Bartonella species, complete genome sequences or assembly contigs of whole genome sequences were aligned by using Mauve version 2.3.1 (22) to identify single nucleotide changes in conserved genomic regions. A total of 12,740 single-nucleotide polymorphisms were found in in 25.2 kb of sequence common to all 38 Bartonella strains. A phylogenetic tree was constructed by using the R phangorn package (23). The initial tree was constructed by using the neighbor-joining algorithm and optimized by using the parsimony maximum-likelihood method. Tree stability was evaluated by using 100 bootstrap replicates.
In the clinical trial testing the efficacy of rifampin and azithromycin for treatment of chronic bartonellosis, blood specimens from 72 of 127 patients were positive for Bartonella species by culture, and gltA gene sequencing indicated that these patients were infected with only B. bacilliformis; however, 2 patients (nos. 20 and 41) were infected with B. ancashensis (17–19). DNA extracts from the 4 original whole blood specimens from these patients (20.00, 20.60, 41.00, and 41.60) were tested by using quantitative bacterial 16S rDNA PCR, standard bacterial 16S PCR, and B. ancashensis–specific PCR (Table 1). Whole blood from patient 20 was PCR negative for bacteria on days 0 and 60. However, high levels of bacteremia were seen for whole blood specimens from days 0 and 60 for patient 41.
Although levels of bacteremia differed greatly, clinical signs and symptoms for both patients were indistinguishable from each another and from those for other patients with confirmed cases of chronic B. bacilliformis infection. In addition, our results confirm that B. ancashensis was isolated from whole blood specimens of patient 41 on day 60, but not on day 0. The evidence suggests emergence of a novel Bartonella species in Peru that can cause its own verruga peruana–like infection in humans or possibly co-infect humans in conjunction with B. bacilliformis. It is intriguing that the bacteremia profile, based on blood cultures, for patient 41 changed from B. bacilliformis at day 0 to B. ancashensis at day 60.
Figure 1

Figure 1. Alignment of whole-genome restriction maps and in silico map for Bartonella isolates from patients with verruga peruana, rural Ancash region, Peru. A) Maps for B. ancashensis isolates 20.00, 20.60, and 41.60...
We performed genome-wide analysis of the 3 B. ancashensis isolates (20.00, 20.60, and 41.60) by using the WGRM and NGS (21,24). The WGRMs of the 3 isolates showed >99.7% similarity with each other and <10% similarity to the WGRM of B. bacilliformis KC583. WGRM showed that the B. ancashensis genome is circular and ≈1.46 Mb. A region of ≈0.64 Mb in the 20.00 genome was inverted when compared with maps for isolates 20.60 and 41.60 (Figure 1).
The complete genome sequence of B. ancashensis strain 20.00 (GenBank accession no. NZ_CP010401) is circular (1,466,048 bp) and has a G + C content of 38.4%. These values are similar to those for the complete genome sequence of B. bacilliformis KC583 (NC_008783.1) (1,445,021 bp and a G + C content of 38.2%). The inverted region of the 20.00 genome (0.64 Mb) is flanked by two 507-bp repetitive sequences in an opposite orientation. The repetitive sequence is 97% identical to the gene for B. bacilliformis integrase (protein family HMM PF00589), a DNA breaking–rejoining enzyme that catalyzes DNA recombination.
Figure 2

Figure 2. Phylogenetic relationship of Bartonella ancashensis isolates from patients with verruga peruana, rural Ancash region, Peru, with other Bartonella species based on whole-genome phylogeny. The tree is based on single-nucleotide polymorphisms identified...
Figure 3
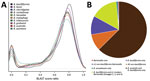
Figure 3. Proteomic analysis of Bartonella ancashensis isolated from patients with verruga peruana, rural Ancash region, Peru, and 10 related Bartonella species. A) Density plot showing similarity between B. ancashensis protein-coding genes and...
We used whole-genome phylogenetic analyses to examine the evolutionary relatedness between B. ancashensis and 40 other Bartonella strains (Table 2; Figure 2). This analysis, based on 12,740 single-nucleotide polymorphisms in genomic sequences conserved across all species, showed that B. ancashensis isolate 20.00 is most closely related to B. bacilliformis, B. bovis, and B. melophagi. The degree of similarity between B. ancashensis and 15 other Bartonella species was further examined by assessing the pairwise similarity of homologous protein with the basic local alignment search tool score ratio method (25) (Figure 3, panel A). B. ancashensis predicted proteins are most similar to B. bacilliformis proteins, but there are several distinctive differences. Specifically, 63.7% of B. ancashensis genes are part of a core genome common to all 15 comparator species, 10.7% of B. ancashensis proteins did not have a homolog in any of the reference Bartonella spp., and 89.3% of B. ancashensis proteins are present in <1 other Bartonella spp.; 79.1% of B. ancashensis proteins were found in B. bacilliformis (Figure 3, panel B).
Most B. ancashensis genes identified by pan-genome analyses had homologs in >1 Bartonella species (Figure 3, panel B). Eight protein-coding genes in B. ancashensis and B. bacilliformis were not present in any other species. An additional 5 protein-coding genes were present in B. ancashensis, B. bacilliformis, and B. australis, but in no other known species. In contrast, there were 8 protein-coding genes in B. ancashensis and all 14 species, excluding B. bacilliformis, but including B. australis, which is considered the most divergent Bartonella species currently recognized. Another 129 B. ancashensis protein-coding genes have homologs in only a subset of the non-bacilliformis Bartonella (26).
Figure 4

Figure 4. Pairwise comparisons of protein-coding genes of Bartonella isolates from patients with verruga peruana, rural Ancash region, Peru. A) Nucleotide similarity of B. ancashensis protein-coding sequences compared with those of B. bacilliformis...
In 3 regions of the B. ancashensis genome, the similarity between B. bacilliformis and B. ancashensis proteins was lower than the average value (Figure 4). Proteins in variable region 1 (genes 30–90) are homologous to non-bacilliformis species proteins, including phage proteins, such as HigA and HigB. Proteins encoded in variable region 2 (genes 180–240) are absent from B. bacilliformis. Among these proteins are those that have high similarity with Bartonella type IV secretion system proteins, which are hypothetical gene products that have moderate identity with proteins from other non-bacilliformis species, and novel hypothetical proteins. Variable region 3 (genes 620–704) contains loci encoding hypothetical proteins not seen in other species and several toxin proteins that are not found in B. bacilliformis, including the RelE/StbE replicon stabilization toxin, the RelB/StbD replicon stabilization protein, and the HigB toxin protein.
Figure 5
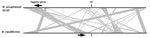
Figure 5. Genetic arrangement of the genome of B. ancashensis isolate 20.00 from a patient with verruga peruana, rural Ancash region, Peru, compared with that of B. bacilliformis KC583. Black lines indicate chromosomes...
Analysis identified 2 characteristic features of B. ancashensis: type IV secretion complex (VirB2) proteins, which are not found in B. bacilliformis; and flagella proteins, which are not found in Bartonella species in lineage 4, including human pathogens B. quintana and B. henselae (15,27,28). In isolates 20.60 and 41.60, the 31 flagellar genes encoded by B. ancashensis are located in the identical order and distances as their homologs in the B. bacilliformis genome. Isolate 20.00 has a large genomic inversion, and this rearrangement results in 1 gene (FliJ) required for production of flagella arranged in a reversed orientation and separated from the other genes of the main flagellar gene cluster. In isolate 20.00, FliJ is ≈600 kb from the flagellar gene cluster; in isolates 20.60 and 41.60, FliJ is ≈100 kb from this cluster (Figure 5).
Figure 6

Figure 6. Virulence-modulating (VM) proteins in Leptospira and Bartonella species. Unrooted phylogenetic tree of VM proteins from Leptospira interrogans (Lin, red), B. australis (Bau, blue), B. bacilliformis (Bbac, green), and B. ancashensis (BAE,...
A Leptospira species virulence attenuation study identified a group of paralogous virulence modulated (VM) genes, which are believed to play a role in human pathogenesis caused by Leptospira interrogans (29). Subsequent comparative genomic analysis showed that VM proteins are present in other bacterial pathogens, including B. bacilliformis and B. australis; B. ancashensis encodes 5 VM proteins (Figure 6). In contrast, no homologs of VM proteins were found by a basic local alignment search tool search in any other recognized Bartonella species. As seen for the VM proteins of Leptospira species, VM protein genes in these 3 Bartonella species were scattered throughout their genomes, and the number of VM proteins was different for each species.
B. bacilliformis was discovered in Peru in 1907 as the causative agent for Oroya fever and verruga peruana. Since then, B. bacilliformis has been the primary subject in bartonelloses studies in South America. However, it has long been speculated that multiple Bartonella spp. might be circulating in this region (30). Our study and previous work clearly indicate B. ancashensis is a unique Bartonella species that co-circulates with B. bacilliformis in the Ancash region of Peru, where the prevalence of B. bacilliformis is high (16,31,32).
Moreover, B. ancashensis has several unique genomic features. Like B. bacilliformis, this species encodes flagellar genes, which are believed to be essential for erythrocyte invasion, the main route of pathogenesis caused by B. bacilliformis (33). In contrast, human pathogens B. quintana and B. henselae do not produce flagella, and host-specific pathogenicity is believed to be linked to the type IV secretion system (T4SS) gene clusters VirB and Trw (27,28,33,34). B. ancashensis and 2 other pathogens (B. clarridgeiae and B. rochalimae) have flagellar and VirB T4SS gene clusters (15,26,27).
Whole-genome analysis of 3 B. ancashensis isolates showed that isolate 20.00 differs from the other 2 isolates by a large genomic region inversion. Future comparative analysis of gene expression profiles in these strains will show whether this genomic inversion alters the regulation of flagellar genes, as well as other virulence factors within or adjacent to the inverted region.
Moreover, it will be useful to investigate whether the genomic inversion (isolate 20.00 vs. 20.60) is related to administration of antimicrobial drugs to infected patients because isolates 20.60 and 41.60 were obtained from patients after they received antimicrobial drugs and cleared any clinical signs or symptoms of bartonellosis because infection persisted 60 days after enrollment in the study and administration of antimicrobial drugs. Furthermore, these isolates were previously tested for their in vitro susceptibility to rifampin and azithromycin and both were found to be susceptible (17). However, in vitro antimicrobial drug susceptibility testing for Bartonella spp. has been largely limited in its clinical utility. However, B. ancashensis might be capable of producing chronic asymptomatic infections that could be caused by its unique genomic characteristics.
The VM genes belong to a family of homologous virulence-related genes originally identified in L. interrogans and modulate the pathogenesis of L. interrogans in humans (29). These genes in B. bacilliformis, B. australis, and B. ancashensis, but not in other Bartonella spp., the further comparative analysis and functional studies on these VM proteins and the large number of other hypothetical proteins in Bartonella spp. will shed light on the pathogenesis mechanisms of bartonellosis, which are so far largely unknown.
Intensified tropical disease surveillance and advances in scientific methods led to an increasing number of new Bartonella species being identified in recent years (30,35). These studies identified 1 major phylogenetic lineage of the genus Bartonella. Our study and other genomic studies demonstrated that B. bacilliformis, which was historically regarded as the ancestral Bartonella spp., probably diverged from other species in the distant past and evolved as a species uniquely adapted to the human host because no small mammals have been implicated as reservoir hosts for B. bacilliformis (15,26,27,36). Although B. ancashensis is a novel species most closely related to B. bacilliformis, it has a nucleotide divergence of ≈20% when compared with B. bacilliformis for conserved genomic regions, which is exceedingly high and comparable with distances among proposed Bartonella lineages (15,26). Therefore, it is rational to designate B. ancashensis as an independent lineage parallel to the B. bacilliformis lineage. Our study provided evidence that there might be more Bartonella species and subspecies in regions of South America.
Bartonellosis has affected humans for hundreds to thousands of years, remains endemic to several areas, and continues to cause sporadic outbreaks in many regions. Identification of a novel Bartonella species in this study not only provided long-awaited evidence of species diversity in areas to which B. bacilliformis is endemic but also indicates the need for acquisition of sufficient genomic data, which will enable pathogenomics studies. Such studies will make essential contribution to a comprehensive understanding and effective control of bartonelloses.
Dr. Mullins is laboratory medicine fellow at the University of Maryland, Baltimore, MD. Her primary research interest is development of infectious disease diagnostic assays.
Acknowledgment
This study was conducted as part of our official duties as employees of the US Government. Therefore, under Title 17 USC paragraph 105, copyright protection is not available.
References
- Breitschwerdt EB, Kordick DL. Bartonella infection in animals: carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clin Microbiol Rev. 2000;13:428–38. DOIPubMedGoogle Scholar
- Harms A, Dehio C. Intruders below the radar: molecular pathogenesis of Bartonella spp. Clin Microbiol Rev. 2012;25:42–78. DOIPubMedGoogle Scholar
- Mosepele M, Mazo D, Cohn J. Bartonella infection in immunocompromised hosts: immunology of vascular infection and vasoproliferation. Clin Dev Immunol. 2012;2012:612809.
- Breitschwerdt EB. Bartonellosis: one health perspectives for an emerging infectious disease. ILAR J. 2014;55:46–58. DOIPubMedGoogle Scholar
- Kosek M, Lavarello R, Gilman RH, Delgado J, Maguiña C, Verástegui M, et al. Natural history of infection with Bartonella bacilliformis in a nonendemic population. J Infect Dis. 2000;182:865–72. DOIPubMedGoogle Scholar
- Rolain JM, Brouqui P, Koehler JE, Maguina C, Dolan MJ, Raoult D. Recommendations for treatment of human infections caused by Bartonella species. Antimicrob Agents Chemother. 2004;48:1921–33. DOIPubMedGoogle Scholar
- Kaiser PO, Riess T, O’Rourke F, Linke D, Kempf VA. Bartonella spp.: throwing light on uncommon human infections. Int J Med Microbiol. 2011;301:7–15. DOIPubMedGoogle Scholar
- Pape M, Kollaras P, Mandraveli K, Tsona A, Metallidis S, Nikolaidis P, et al. Occurrence of Bartonella henselae and Bartonella quintana among human immunodeficiency virus-infected patients. Ann N Y Acad Sci. 2005;1063:299–301. DOIPubMedGoogle Scholar
- Chaloner GL, Harrison TG, Birtles RJ. Bartonella species as a cause of infective endocarditis in the UK. Epidemiol Infect. 2013;141:841–6. DOIPubMedGoogle Scholar
- Raoult D, Fournier PE, Drancourt M, Marrie TJ, Etienne J, Cosserat J, et al. Diagnosis of 22 new cases of Bartonella endocarditis. Ann Intern Med. 1996;125:646–52. DOIPubMedGoogle Scholar
- Foucault C, Barrau K, Brouqui P, Raoult D. Bartonella quintana bacteremia among homeless people. Clin Infect Dis. 2002;35:684–9. DOIPubMedGoogle Scholar
- Drali R, Sangaré AK, Boutellis A, Angelakis E, Veracx A, Socolovschi C, et al. Bartonella quintana in body lice from scalp hair of homeless persons, France. Emerg Infect Dis. 2014;20:907–8. DOIPubMedGoogle Scholar
- Birtles RJ, Fry NK, Ventosilla P, Cáceres AG, Sánchez E, Vizcarra H, et al. Identification of Bartonella bacilliformis genotypes and their relevance to epidemiological investigations of human bartonellosis. J Clin Microbiol. 2002;40:3606–12. DOIPubMedGoogle Scholar
- Buffet JP, Kosoy M, Vayssier-Taussat M. Natural history of Bartonella-infecting rodents in light of new knowledge on genomics, diversity and evolution. Future Microbiol. 2013;8:1117–28. DOIPubMedGoogle Scholar
- Engel P, Salzburger W, Liesch M, Chang C-C, Maruyama S, Lanz C, et al. Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella. PLoS Genet. 2011;7:e1001296. DOIPubMedGoogle Scholar
- Sanchez Clemente N, Ugarte-Gil CA, Solórzano N, Maguiña C, Pachas P, Blazes D, et al. Bartonella bacilliformis: a systematic review of the literature to guide the research agenda for elimination. PLoS Negl Trop Dis. 2012;6:e1819. DOIPubMedGoogle Scholar
- Mullins KE, Hang J, Jiang J, Leguia M, Kasper MR, Ventosilla P, et al. Description of Bartonella ancashensis sp. nov., isolated from the blood of two patients with verruga peruana. Int J Syst Evol Microbiol. 2015;65:3339–43. DOIPubMedGoogle Scholar
- Blazes DL, Mullins K, Smoak BL, Jiang J, Canal E, Solorzano N, et al. Novel Bartonella agent as cause of verruga peruana. Emerg Infect Dis. 2013;19:1111–4. DOIPubMedGoogle Scholar
- Mullins KE, Hang J, Jiang J, Leguia M, Kasper MR, Maguiña C, et al. Molecular typing of “Candidatus Bartonella ancashi,” a new human pathogen causing verruga peruana. J Clin Microbiol. 2013;51:3865–8. DOIPubMedGoogle Scholar
- Hang J, Mullins KE, Clifford RJ, Onmus-Leone F, Yang Y, Jiang J, et al. Complete genome sequence of Bartonella ancashensis strain 20.00, isolated from the blood of a patient with verruga peruana. Genome Announc. 2015;3:3. DOIPubMedGoogle Scholar
- Onmus-Leone F, Hang J, Clifford RJ, Yang Y, Riley MC, Kuschner RA, et al. Enhanced de novo assembly of high throughput pyrosequencing data using whole genome mapping. PLoS One. 2013;8:e61762. DOIPubMedGoogle Scholar
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. DOIPubMedGoogle Scholar
- Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3. DOIPubMedGoogle Scholar
- Riley MC, Kirkup BC Jr, Johnson JD, Lesho EP, Ockenhouse CF. Rapid whole genome optical mapping of Plasmodium falciparum. Malar J. 2011;10:252. DOIPubMedGoogle Scholar
- Lerat E, Daubin V, Moran NA. From gene trees to organismal phylogeny in prokaryotes: the case of the γ-Proteobacteria. PLoS Biol. 2003;1:E19. DOIPubMedGoogle Scholar
- Guy L, Nystedt B, Toft C, Zaremba-Niedzwiedzka K, Berglund EC, Granberg F, et al. A gene transfer agent and a dynamic repertoire of secretion systems hold the keys to the explosive radiation of the emerging pathogen Bartonella. PLoS Genet. 2013;9:e1003393. DOIPubMedGoogle Scholar
- Dehio C. Infection-associated type IV secretion systems of Bartonella and their diverse roles in host cell interaction. Cell Microbiol. 2008;10:1591–8. DOIPubMedGoogle Scholar
- Eicher SC, Dehio C. Bartonella entry mechanisms into mammalian host cells. Cell Microbiol. 2012;14:1166–73. DOIPubMedGoogle Scholar
- Lehmann JS, Fouts DE, Haft DH, Cannella AP, Ricaldi JN, Brinkac L, et al. Pathogenomic inference of virulence-associated genes in Leptospira interrogans. PLoS Negl Trop Dis. 2013;7:e2468. DOIPubMedGoogle Scholar
- Chaloner GL, Palmira Ventosilla , Birtles RJ. Multi-locus sequence analysis reveals profound genetic diversity among isolates of the human pathogen Bartonella bacilliformis. PLoS Negl Trop Dis. 2011;5:e1248. DOIPubMedGoogle Scholar
- Chamberlin J, Laughlin LW, Romero S, Solórzano N, Gordon S, Andre RG, et al. Epidemiology of endemic Bartonella bacilliformis: a prospective cohort study in a Peruvian mountain valley community. J Infect Dis. 2002;186:983–90. DOIPubMedGoogle Scholar
- Chamberlin J, Laughlin L, Gordon S, Romero S, Solórzano N, Regnery RL. Serodiagnosis of Bartonella bacilliformis infection by indirect fluorescence antibody assay: test development and application to a population in an area of bartonellosis endemicity. J Clin Microbiol. 2000;38:4269–71.PubMedGoogle Scholar
- Scherer DC, DeBuron-Connors I, Minnick MF. Characterization of Bartonella bacilliformis flagella and effect of antiflagellin antibodies on invasion of human erythrocytes. Infect Immun. 1993;61:4962–71.PubMedGoogle Scholar
- Deng HK, Le Rhun D, Le Naour E, Bonnet S, Vayssier-Taussat M. Identification of Bartonella Trw host-specific receptor on erythrocytes. PLoS One. 2012;7:e41447. DOIPubMedGoogle Scholar
- Eremeeva ME, Gerns HL, Lydy SL, Goo JS, Ryan ET, Mathew SS, et al. Bacteremia, fever, and splenomegaly caused by a newly recognized bartonella species. N Engl J Med. 2007;356:2381–7. DOIPubMedGoogle Scholar
- Minnick MF, Battisti JM. Pestilence, persistence and pathogenicity: infection strategies of Bartonella. Future Microbiol. 2009;4:743–58. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1Current affiliation: University of Maryland, Baltimore, Maryland, USA.
2These authors contributed equally to this article.
Table of Contents – Volume 23, Number 3—March 2017
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Kristin E. Mullins, Department of Pathology, School of Medicine, University of Maryland, Baltimore, MD 21210, USA
Top