Volume 24, Number 5—May 2018
Research
Epidemic Dynamics of Vibrio parahaemolyticus Illness in a Hotspot of Disease Emergence, Galicia, Spain
Cite This Article
Citation for Media
Abstract
Galicia in northwestern Spain has been considered a hotspot for Vibrio parahaemolyticus infections. Infections abruptly emerged in 1998 and, over the next 15 years, were associated with large outbreaks caused by strains belonging to a single clone. We report a recent transition in the epidemiologic pattern in which cases throughout the region have been linked to different and unrelated strains. Global genome-wide phylogenetic analysis revealed that most of the pathogenic strains isolated from infections were associated with globally diverse isolates, indicating frequent episodic introductions from disparate and remote sources. Moreover, we identified that the 2 major switches in the epidemic dynamics of V. parahaemolyticus in the regions, the emergence of cases and an epidemiologic shift in 2015–2016, were associated with the rise of sea surface temperature in coastal areas of Galicia. This association may represent a fundamental contributing factor in the emergence of illness linked to these introduced pathogenic strains.
Globally, Vibrio parahaemolyticus is the leading bacteriological cause of illness associated with seafood consumption. Infections have undergone a global expansion over the last 2 decades; cases have suddenly emerged in areas considered environmentally adverse for these pathogens (1–3). The causes of this dynamic expansion and emergence in non–disease-endemic areas have remained elusive.
V. parahaemolyticus infections are generally rare and sporadic across all of Europe with a single exception: Galicia in northwestern Spain. This region has been considered a hotspot for Vibrio infections and an anomaly within the epidemiologic context of V. parahaemolyticus in Europe; reccurring cases of foodborne vibriosis (4–7) and outbreaks (7,8) have been reported regularly since the late 1990s. Infections associated with V. parahaemolyticus in Galicia were characterized by sudden outbreaks of illness typically associated with a single genetic variant of the pathogen (6,8). The first sign of change in this epidemiologic pattern was observed in 2012 when 3 different and genetically unrelated strains of V. parahaemolyticus were identified during a large outbreak in Galicia (5,9). Since then, a clear transition in the epidemiology of this pathogen has been observed; sporadic cases scattered throughout the region have been caused by different and unrelated strains and typically associated with the consumption of locally produced shellfish.
We applied whole-genome sequencing for a comprehensive and high-resolution insight into pathogenic populations identified in clinical sources associated with the major episodes of illness in Galicia over the past 20 years. We performed phylogenetic analysis to identify the population structure and potential sources of the clinical strains identified in Galicia. We also conducted a parallel exploration of the variability of environmental conditions in the region to investigate the existence of other factors contributing to the emergence of illness linked to these in this particular area.
Bacterial Strains and DNA Extraction
We analyzed 18 isolates derived from clinical sources collected over the course of the different outbreaks in Galicia (Table). All the strains isolated from infections in Galicia were characteristically tdh positive and trh negative, the only exceptions being the strains belonging to sequence type (ST) 36 isolated during the 2012 outbreak, which were positive for both haemolysin genes. Additionally, we included 14 isolates obtained from environmental sources (shellfish and zooplankton) during 2003–2007 to analyze the potential connection between the clinical pathogenic populations and local marine environmental sources. Finally, we added 4 clinical strains reported in the United Kingdom associated with human infections since the 1970s to the study to explore possible connections between pathogenic populations within Europe, along with another 6 environmental strains from the United Kingdom isolated in 2014.
Genome Sequencing and Sequence Processing
We performed genomic DNA extraction of the 42 strains from overnight cultures using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany). We sequenced the genomes of all 42 strains using MiSeq (Illumina, San Diego, CA, USA) with a minimum coverage of 40–120-fold. We prepared libraries with the Nextera XT DNA sample preparation kit (Illumina) and de novo assembled whole-genome sequence contigs for each strain by using CLC Genomics Workbench version 7.5.1 (QIAGEN, Valencia, CA, USA).
Global Collection of V. parahaemolyticus Genomes
We initially investigated the position of the strains from Spain and the United Kingdom on the global phylogeny of V. parahaemolyticus using all the available V. parahaemolyticus genomes worldwide, including 696 genomes obtained from the National Center for Biotechnology Information Assembly and Sequence Read Archive (SRA) databases (Technical Appendix Table) plus the 42 genomes sequenced in our study. We transformed the SRA data to fastq using SRA Toolkit (fastq-dump –split-files –gzip –skip-technical) (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc). We performed genome assembly with A5-pipeline (10). We performed in silico inference of MLST profiles and STs using MLST software (https://github.com/tseemann/mlst), which infers STs using the public MLST scheme for V. parahaemolyticus based on 7 housekeeping genes (https://pubmlst.org/vparahaemolyticus/).
Single-Nucleotide Polymorphism Calling and Phylogenetic Inference
Single-nucleotide polymorphisms (SNPs) were called using Harvest version 1.0.1 (https://github.com/marbl/harvest) (11). We used Parsnp, a component of the Harvest suite, to align the assembled genomes and define the core genome. We identified SNPs for both chromosomes by Parsnp in the multi-alignments; we used filtered and reliable core-genome SNPs to construct a core genome maximum-likelihood phylogenetic tree.
Analysis of Sea Surface Temperature Trend Off the Coast of Galicia, Northwest of Spain
We estimated trend in the mean values of sea surface temperature (SST) using daily SST data from a coastal area limited by the coordinates 42°N–43°N and 8.5°W–9.5°W. The mean SST data come from the Optimum Interpolation SST 1/4° daily dataset (OISST), which extends from September 1981 to the present and is distributed by NOAA/NCEI. This dataset combines satellite retrievals and in situ SST data from ships and buoys. We use these analyzed fields to estimate the trends in the region of interest, detect possible regime shifts, and study the habitat suitability of Vibrio spp. in the region. We investigated regime shift, defined as rapid reorganizations of ecosystems from one relatively stable state to another, using Sequential Regime Shift Detection Software version 3.2 (http://www.beringclimate.noaa.gov/regimes/). This program detects statistically significant shifts in the mean level and magnitude of fluctuations in time series taking the autocorrelation into account (12). The program detects shifts in the mean level of SSTs. The method is based on a sequential t-test that can signal a possibility of a regime shift. We used the default significance level of 0.1 that represents the level at which the null hypothesis that the mean values of the 2 regimes are equal is rejected by the t-test.
Nucleotide Sequence Accession Numbers
The draft genome sequences of all 44 V. parahaemolyticus strains from our study are available in GenBank. Accession numbers are provided in the Table.
Figure 1
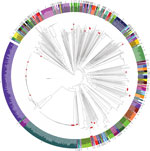
Figure 1. Phylogenetic reconstruction of Vibrio parahaemolyticus based on 738 available genomes. Red dots indicate isolates from Spain collected over the past 20 years from clinical settings and environmental sources. Colors represent sequence...
Figure 2

Figure 2. Phylogeny of Vibrio parahaemolyticus isolates from Galicia, Spain. A) Phylogenetic inference of the 42 genomes from Spain identified in this study (red text) along with all other genomes identified in the...
Analysis of the 738 V. parahaemolyticus genomes resulted in a core genome alignment of 292,750 bp containing 12,399 SNPs. Positions of the Spanish strains in the global phylogeny (Figure 1) revealed a complex epidemiologic scenario with the existence of multiple, highly diverse genomic variants of strains associated with infections in the region. Moreover, we identified 12 different STs among clinical strains isolated over the past 2 decades (Figures 1, 2). We selected the genomes that clustered together with the genomes from Spain (115 genomes) and included them in a high-resolution phylogenetic reconstruction (Figure 2). The basis for the reconstruction was a core genome alignment of 3,049,195 bp containing 202,859 SNPs.
The original strain that was isolated over the course of the earliest documented large outbreak in Galicia in 1998–1999 (ST17), which pulsed-field gel electrophoresis (PFGE) subsequently reported as a new local clone (6), turned out to be closely related to strains previously isolated in Thailand (2006) and the United States (2006) when assessed by whole-genome phylogeny. The clinical strain from the United Kingdom isolated in Maidstone in the late 1970s (National Collection of Type Cultures no. 11344) and reported as genetically related to the Galician strains by PFGE (6), was also found to cluster with this group, with a difference of 450 SNPs.
Strains belonging to the so-called Asian pandemic clone (clonal complex [CC] 3) were first reported in Galicia in association with a large outbreak in 2004 (8). Epidemiologic analysis of the outbreak traced back the origin of the outbreak to a facility located in the international seaport of A Coruña in Galicia, suggesting that the most probable source of the pandemic strain was the discharge of ballast water carried in ships. No strain from this group was identified in Spain until summer 2016, when we identified 2 strains isolated in July from 2 independent outbreaks in the cities of Silleda and Pontevedra, investigated in 2 different hospitals, as belonging to CC3. Genomic analysis of the strains from Galicia, along with 21 additional genomes belonging to the ST3 strains isolated in other countries, resulted in a core alignment of 3,560,214 bp for the ST3 clade containing 384 SNPs. Whole-genome phylogeny revealed that the 2 strains from Galicia isolated in 2016 belonged to 2 different groups; both are different from the strain identified in Galicia in 2004. We identified strain N314 as part of the Asian group of CC3 and strain CFSAN056086 in the American group. We were able to clearly distinguish these 2 strains from the strains isolated from the 2004 outbreak, which was identified in a separate branch closer to different Asian strains. Of note, while the 2004 outbreak was associated with imported seafood and unsafe food manipulation (8), the recent infections caused by CC3 strains were unequivocally associated with local shellfish (razor clams and cockles); compelling evidence of several successful introduction events of these strains into the marine environment of Galicia.
In many ways, the 2012 outbreak in Spain (5,13) represented a clear change in the epidemic dynamics of V. parahaemolyticus in the region. First, this outbreak was the largest reported across Europe linked to local seafood; second, it was the earliest known evidence of a cross-continental spreading of the ST36 clone, which is endemic to the Pacific Northwest (PNW) of the United States and one of the most virulent ST groups (13). Genomic analysis of the 92 available genomes of the ST36 isolated from areas of endemicity for this group in the PNW identified a core alignment of 3,310,986 bp comprising 1,596 SNPs. Phylogenetic analysis of the ST36 lineage revealed the existence of 2 different clusters within this group (Figure 2, panel C): a first cluster composed of old strains from Canada isolated before 2005 and the United States before 2000, and a second cluster with modern representatives from the United States and Canada. This particular population structure suggests the existence of a lineage replacement in the PNW coast and western Canada, where only strains belonging to the second cluster were identified from 2005 onward. Surprisingly, we unequivocally identified the strains in the 2012 Galicia outbreak as belonging to the first cluster composed of strains extinct in their original location along the PNW coast, which suggests an early introduction of these strains into waters of Galicia and Europe (14). Furthermore, we identified a single strain from Canada as closely related to the genomes from the Galician strains with a minimum difference of 20–22 SNPs in an alignment of 3,310,986 bp, whereas variations among genomes of the ST36 strains from Galicia were 0–19 SNPs. The low level of variation among all the genomes in this clade supports a hypothesis that Galician strains originated in British Columbia, Canada, and were introduced in Galicia sometime after 2001.
A second singularity of the 2012 outbreak, and probably more noteworthy, was the fact that infections from a single outbreak were associated with several unrelated strains of V. parahaemolyticus. We identified 2 additional strains different from ST36 from clinical cases over the course of this outbreak, ST1031 and ST1032; both represent novel STs not reported before the 2012 outbreak. Whole-genome phylogeny of these new STs grouped these strains into 2 distinctive clusters. Strain G32, belonging to ST1032, showed a close association with several strains isolated from zooplankton in offshore waters of Galicia in 2006–2007, which could be preliminary evidence of a local origin of these strains introduced by the incursion of offshore oceanic waters. Strain G33, belonging to ST1031, was included in a single group along with strains also associated with local shellfish and isolated over the course of the outbreaks in summers of 2015 and 2016.
Finally, we identified an additional group of strains belonging to ST327 associated with illnesses over the summer of 2015 and 2016. We included these strains in the same cluster as 1 strain from Thailand isolated in 1990 (Figure 2).
Our genome-wide phylogenetic analysis of pathogenic V. parahaemolyticus in northern Spain has provided novel insights into the epidemiology of V. parahaemolyticus in nonendemic areas. The primary result is that the study revealed the existence of a complex epidemiologic context characterized by the existence of multiple highly diverse strains, most originating far away, that caused infections associated with locally produced shellfish; this finding could be considered evidence of multiple events of introduction of foreign variants into Galicia. The source of these strains into Galicia is elusive and remains an area of ongoing interest, but we did identify through this study and previous work the 2 proposed mechanisms for dissemination of pathogenic strains: ballast water (15–17) and zooplankton migration (18,19). Ballast water has been proposed as a main source of pathogenic Vibrio bacteria (16) and was suggested as the mechanism of introduction of pandemic strains in the 2004 outbreak (8). In addition, we showed new evidence that unequivocally identified environmental transport through offshore zooplankton as one of the routes of introduction of new pathogenic variants via ocean currents (18–20). However, we cannot rule out the introduction of foreign mollusks into the marine water of Galicia as a possible source of new variants of pathogenic Vibrio bacteria from disparate and remote sources because of the magnitude of the shellfish trade in the region; the importation of shellfish from other geographic areas is a common practice to supply the high demand for products. A recent study analyzing the population structure and evolution of the ST36 clone suggests that the importation of clams from the PNW to Spain circa 2000 is the probable source of ST36 strains (14). Moreover, 2 other recent studies tracking the routes of introduction of the Manila clam from its original place of distribution in the Indo-Pacific region to Europe has also linked the origin of clam populations introduced in Spain to the PNW of the United States (area of endemicity of ST36 populations) during the importations of clams in the mid- and late 1990s (21,22). These findings closely correspond with the results shown in our study.
Figure 3
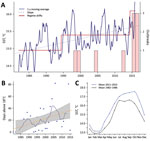
Figure 3. Recent environmental warming trends in Galicia, Spain, 1982–2016. Trends in the mean values of SST were estimated using daily SST data from a coastal area defined by the coordinates 42°–43°N and...
An underlying finding of our study is that the introduction of these highly pathogenic strains into a region is not sufficient by itself to initiate an epidemic; the introduced strains appear established in the area for a substantial period without evidence of associated illness, which suggests additional cofactors in infections and risk. Because seawater temperature has been identified as a critical factor governing the emergence of Vibrio diseases (1–3), we conducted an analysis of historical records of SST in the region. Results from these analyses revealed a significant trend of increased SST in the area, which followed a stepwise and incremental trend, rather than the expected linear change. We identified shifts in both the mean level of fluctuations and the variance of SST time series. We recognized 2 clear shifts of SST with a significant statistical support over the study period: June 1994, an SST increase of 0.4°C; and June 2014, an SST increase of 0.7°C (Figure 3). These 2 shifts in seawater temperature showed a close correspondence with the epidemic dynamics of V. parahaemolyticus in the area, showing a period with no infections before the first regime shift, a second period when the first epidemic events were identified, and finally a third period after the last regime shift in 2014, which was concurrent with the change in the epidemiology of V. parahaemolyticus we report in this study. Previous data have shown that regime shift warming has led to an increase in prevalence of Vibrio bacteria in the environment (1,23), and epidemiologic studies on the emergence of Vibrio infections have identified SSTs >18°C as a critical threshold for triggering infections and substantially increasing the number of reported clinical cases (2). Analysis of the number of days with SSTs >18°C in Galicia over the past 36 years identified an increase of 1 day/year (Figure 2, panel B), which resulted in an increase of 35 days for the risk period of V. parahaemolyticus infections for the whole period. These results contrast with the situation in some areas of natural endemicity for pathogenic V. parahaemolyticus, such as the PNW for the ST36, where seawater temperatures have remained remarkably stable over the past 2 decades and regime shifts have not been detectable. The average annual SST in areas of Puget Sound in the PNW is ≈5°C lower than in Galicia, showing a warming trend almost 2 orders of magnitude smaller and SST values always <18°C, according to the records of the satellite mesoscale SST time series (data not shown).
This study highlights the utility of whole-genome sequencing as a tool to elucidate key features of the transmission and potential sources of pathogenic environmental bacteria such as Vibrio spp. The concomitant introduction of foreign Vibrio variants with a significant warming trend in the region, coupled with the consumption of locally produced shellfish in the region, may be major contributing factors for the emergence of infections in Galicia. Parallel circumstances may also drive disease emergence in other areas of the world with similar environmental conditions, such as the Pacific Northwest (24,25) and the Atlantic Northeast (14,26) in the United States or the south of Chile (27). In these areas. The presence of imported Vibrio strains has been frequently reported associated with outbreaks, particularly during and after warming events (28). These areas represent major contributors to the escalation and global expansion of V. parahaemolyticus illnesses associated with the dissemination of the preeminent pathogenic clones of these organisms.
Dr. Martinez-Urtaza is a senior scientist at the Centre for Environment, Fisheries and Aquaculture Science in Weymouth, United Kingdom. His work covers aspects of molecular epidemiology and effects of climate on infectious diseases with research interests focused on the study of foodborne and waterborne diseases.
Acknowledgment
J.T. was funded by National Oceanic and Atmospheric Administration (NOAA)/OceanWatch and NOAA/Atlantic Oceanographic and Meteorological Laboratory. M.A. was funded through the NERC project NE/P004121/1.
References
- Vezzulli L, Grande C, Reid PC, Hélaouët P, Edwards M, Höfle MG, et al. Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc Natl Acad Sci U S A. 2016;113:E5062–71. DOIPubMedGoogle Scholar
- Baker-Austin CL, Trinanes JA, Taylor NGH, Hartnell R, Siitonen A, Martinez-Urtaza J. Emerging Vibrio risk at high latitudes in response to ocean warming. Nat Clim Chang. 2012;3:73–7. DOIGoogle Scholar
- Martinez-Urtaza J, Bowers JC, Trinanes J, DePaola A. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res Int. 2010;43:1780–90. DOIGoogle Scholar
- Martinez-Urtaza J, Lozano-Leon A, Varela-Pet J, Trinanes J, Pazos Y, Garcia-Martin O. Environmental determinants of the occurrence and distribution of Vibrio parahaemolyticus in the rias of Galicia, Spain. Appl Environ Microbiol. 2008;74:265–74. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Powell A, Jansa J, Rey JL, Montero OP, Campello MG, et al. Epidemiological investigation of a foodborne outbreak in Spain associated with U.S. West Coast genotypes of Vibrio parahaemolyticus. Springerplus. 2016;5:87. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Lozano-Leon A, DePaola A, Ishibashi M, Shimada K, Nishibuchi M, et al. Characterization of pathogenic Vibrio parahaemolyticus isolates from clinical sources in Spain and comparison with Asian and North American pandemic isolates. J Clin Microbiol. 2004;42:4672–8. DOIPubMedGoogle Scholar
- Lozano-León A, Torres J, Osorio CR, Martínez-Urtaza J. Identification of tdh-positive Vibrio parahaemolyticus from an outbreak associated with raw oyster consumption in Spain. FEMS Microbiol Lett. 2003;226:281–4. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Simental L, Velasco D, DePaola A, Ishibashi M, Nakaguchi Y, et al. Pandemic Vibrio parahaemolyticus O3:K6, Europe. Emerg Infect Dis. 2005;11:1319–20. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Baker-Austin C, Jones JL, Newton AE, Gonzalez-Aviles GD, DePaola A. Spread of Pacific Northwest Vibrio parahaemolyticus strain. N Engl J Med. 2013;369:1573–4. DOIPubMedGoogle Scholar
- Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31:587–9. DOIPubMedGoogle Scholar
- Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. DOIPubMedGoogle Scholar
- Rodionov SN. Use of prewhitening in climate regime shift detection. Geophys Res Lett. 2006;33:L12707. DOIGoogle Scholar
- Martinez-Urtaza J, Baker-Austin C, Jones JL, Newton AE, Gonzalez-Aviles GD, DePaola A. Spread of Pacific Northwest Vibrio parahaemolyticus strain. N Engl J Med. 2013;369:1573–4. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, van Aerle R, Abanto M, Haendiges J, Myers RA, Trinanes J, et al. Genomic variation and evolution of Vibrio parahaemolyticus ST36 over the course of a transcontinental epidemic expansion. MBio. 2017;8:e01425–17. DOIPubMedGoogle Scholar
- McCarthy SA, Khambaty FM. International dissemination of epidemic Vibrio cholerae by cargo ship ballast and other nonpotable waters. Appl Environ Microbiol. 1994;60:2597–601.PubMedGoogle Scholar
- DePaola A, Capers GM, Motes ML, Olsvik O, Fields PI, Wells J, et al. Isolation of Latin American epidemic strain of Vibrio cholerae O1 from US Gulf Coast. Lancet. 1992;339:624. DOIPubMedGoogle Scholar
- Ruiz GM, Rawlings TK, Dobbs FC, Drake LA, Mullady T, Huq A, et al. Global spread of microorganisms by ships. Nature. 2000;408:49–50. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Blanco-Abad V, Rodriguez-Castro A, Ansede-Bermejo J, Miranda A, Rodriguez-Alvarez MX. Ecological determinants of the occurrence and dynamics of Vibrio parahaemolyticus in offshore areas. ISME J. 2012;6:994–1006. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Trinanes J, Gonzalez-Escalona N, Baker-Austin C. Is El Niño a long-distance corridor for waterborne disease? Nat Microbiol. 2016;1:16018. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Huapaya B, Gavilan RG, Blanco-Abad V, Ansede-Bermejo J, Cadarso-Suarez C, et al. Emergence of Asiatic Vibrio diseases in South America in phase with El Niño. Epidemiology. 2008;19:829–37. DOIPubMedGoogle Scholar
- Chiesa S, Lucentini L, Freitas R, Nonnis Marzano F, Breda S, Figueira E, et al. A history of invasion: COI phylogeny of Manila clam Ruditapes philippinarum in Europe. Fish Res. 2017;186:25–35. DOIGoogle Scholar
- Cordero D, Delgado M, Liu B, Ruesink J, Saavedra C. Population genetics of the Manila clam (Ruditapes philippinarum) introduced in North America and Europe. Sci Rep. 2017;7:39745. DOIPubMedGoogle Scholar
- Vezzulli L, Brettar I, Pezzati E, Reid PC, Colwell RR, Höfle MG, et al. Long-term effects of ocean warming on the prokaryotic community: evidence from the vibrios. ISME J. 2012;6:21–30. DOIPubMedGoogle Scholar
- Paranjpye R, Hamel OS, Stojanovski A, Liermann M. Genetic diversity of clinical and environmental Vibrio parahaemolyticus strains from the Pacific Northwest. Appl Environ Microbiol. 2012;78:8631–8. DOIPubMedGoogle Scholar
- Turner JW, Paranjpye RN, Landis ED, Biryukov SV, González-Escalona N, Nilsson WB, et al. Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS One. 2013;8:e55726. DOIPubMedGoogle Scholar
- Newton AE, Garrett N, Stroika SG, Halpin JL, Turnsek M, Mody RK; Centers for Disease Control and Prevention (CDC). Increase in Vibrio parahaemolyticus infections associated with consumption of Atlantic Coast shellfish—2013. MMWR Morb Mortal Wkly Rep. 2014;63:335–6.PubMedGoogle Scholar
- González-Escalona N, Cachicas V, Acevedo C, Rioseco ML, Vergara JA, Cabello F, et al. Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg Infect Dis. 2005;11:129–31. DOIPubMedGoogle Scholar
- Raszl SM, Froelich BA, Vieira CR, Blackwood AD, Noble RT. Vibrio parahaemolyticus and Vibrio vulnificus in South America: water, seafood and human infections. J Appl Microbiol. 2016;121:1201–22. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 24, Number 5—May 2018
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jaime Martinez-Urtaza, The Centre for Environment, Fisheries and Aquaculture Science, The Nothe, Barrack Road, Weymouth, Dorset DT4 8UB, UK
Top