Volume 26, Number 8—August 2020
Dispatch
SARS-CoV-2 Phylogenetic Analysis, Lazio Region, Italy, February–March 2020
Cite This Article
Citation for Media
Abstract
We report phylogenetic and mutational analysis of severe acute respiratory syndrome coronavirus 2 virus strains from the Lazio region of Italy and provide information about the dynamics of virus spread. Data suggest effective containment of clade V strains, but subsequently, multiple waves of clade G strains were circulating widely in Europe.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has raised serious concerns because of its rapid dissemination worldwide. Italy is one of the countries with the highest number of coronavirus disease (COVID-19) cases (1,2). Nevertheless, the information about the molecular epidemiology of SARS-CoV-2 strains circulating in Italy is still limited. The analysis of sequence data shown in GISAID (https://www.gisaid.org) indicates that the initial introduction of SARS-CoV-2 in Italy through 2 infected tourists in January was effectively contained (3), and no further circulation of similar clade V strains has been so far detected. An intense wave of infections occurred afterwards, initially affecting Lombardy and Veneto and later on all the other regions of Italy. The strains detected in Italy since February 20 belonged only to clade G. This clade, apparently originating in Shanghai, has been widely circulating in the European Union (EU) countries before reaching Italy (3–5).
Preliminary data suggested that multiple introductions of clade G strains have occurred in Italy, giving rise to contemporary circulation of different strains also detected in other EU countries; this pattern suggests that, after partially undetected introduction of the virus in EU from China, the circulation of travelers within EU ignited virus spread in Europe. We report the phylogenetic and mutational analysis of SARS-CoV-2 strains detected in the Lazio region of Italy, providing additional information on the dynamics of virus dissemination in this country.
We analyzed nasopharyngeal swab (n = 6) and bronchoalveolar lavage (n = 3) samples from 9 patients with COVID-19 to perform SARS-CoV-2 whole-genome reconstruction and mutational analysis. We collected samples in late February and early March, 2020 (Table 1). At sampling time, all patients reported symptoms such as fever, sore throat, cough, or other respiratory symptoms. Two sequences were identical, so we included only 1 of them in the analysis, resulting in 8 total sequences. We named the sequences INMI3–INMI10 for their detection at National Institute for Infectious Diseases and analyzed them together with the previously published INMI1 and INMI2 (6), along with all the sequences from Italy posted to GISAID database by April 11, 2020.
We performed next-generation sequencing (SARS-CoV-2 Panel) on Ion Torrent platform (Thermo Fisher Scientific, https://www.thermofisher.com) using shotgun approach for INMI3–4 and amplicon approach for INMI5–10. After quality control, we generated a median number of 4.3 × 107 reads for each shotgun sample and 1.5 × 106 for each amplicon sample (ranging from 7.5 × 105 to 4.8 × 107). The sequencing mean depth of SARS-CoV-2 ranged from 367-fold in INMI3 to 16,661-fold in INMI5.
Figure
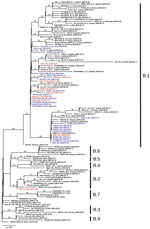
Figure. Phylogenetic analysis of 150 severe acute respiratory syndrome coronavirus 2 representative genome sequences, including genomes collected in Italy (blue) and sequences identified for this study at the National Institute for Infectious...
We submitted consensus sequences to GISAID. We used the proposed phylogenetic lineage classification (A. Rambaut et al., unpub. data, https://doi.org/10.1101/2020.04.17.046086) in phylogenetic analysis; for comparison to previously published reports, we maintained references to clades reported in GISAID. INMI1 and INMI2 are included in clade V according to GISAID phylogenetics, as reported (6), and clade B2; the clade includes other sequences from EU countries, but no additional sequences from Italy. All other INMI sequences cluster with the GISAID G clade, and with the B1 clade; we focused subsequent analysis on clade B1 (Figure).
The clade B1 INMI sequences are distributed in 2 main clusters, one including most of the northern Italy strains and the other including sequences mainly from central Italy. In particular, INMI4, which was epidemiologically linked to Bergamo (Lombardy region), clusters with sequences from central Italy (Abruzzo region). The other INMI sequences cluster with strains from northern Italy. Of note, in both clusters the sequences from Italy are intermixed with sequences from other EU countries, which can also be seen in the broader phylogenetic analysis on GISAID, in which more EU sequences are analyzed. We have identified 5 synonymous and 9 nonsynonymous substitutions distributed along the whole genome (Table 2).
Each patient showed several amino acid substitutions ranging from 4 to 7. The G clade–specific single-nucleotide polymorphism A23403G led the amino acid change D614G in the S protein. We observed one additional mutation in this protein, that of C21575T (L5F) in INMI7, which is detected in few other sequences in GISAID, interspersed among different non-G clades (M. Chiara et al., unpub. data, https://doi.org/10.1101/2020.03.30.016790). Its location in a marginal region of the gene and the sporadic distribution in different clades indicates repeated occurrence not followed by fixation, consistent with no evolutionary advantage.
The S protein in the SARS-CoV-2 virus is a chief determinant of the host range and pathogenicity. The virion attaches to the cell membrane by binding the S protein with the host ACE2 receptor (7). The D614G mutation, located in the putative S1–S2 junction region near the furin polybasic cleavage site (RRAR), might have an effect on priming by host cell proteases; however, the real impact of this high-frequency mutation is unclear.
The variants C241T, C3037T (located in the noncoding region) and C14408T (in open reading frame1ab, orf1ab) were present in all INMI3–INMI10 sequences. These mutations have been detected in several SARS-CoV-2 isolates throughout Europe and are characteristic of clade G (C. Yin, unpub. data). A nonsynonymous substitution D3G in membrane glycoprotein was detected in 1 INMI9 sequence.
We detected 3 nucleotide changes in INMI4, located in a high variable region of the gene, in 2 adjacent codons of the nucleocapsid (N) gene, two 2-amino acid changes, R203K and G204R. N protein, responsible for the formation of helical nucleocapsid, can elicit humoral and cell mediated immune response and has potential value in vaccine development. However, none of the observed mutations has been so far associated with changes in viral pathogenicity or transmissibility.
The phylogenetic reconstruction we report suggests possible multiple introduction of SARS-CoV-2 virus in Italy, supporting previously reported analysis conducted on a more limited number of sequences (3–5).
The analysis consistently places the strains described in this study in 2 distinct clusters in B1 clade. No other sequence from Italy clusters in B2 (or GISAID V) clade, indicating the positive effect of containment measures established by health authorities in both Italy and China to limit viral transmission directly from China. The same measures were unable to contain a wave of subsequent multiple introductions in Italy of strains that were widely circulating in Europe, all clustering with clade B1.
The inclusion of the viral sequences from infections occurring in the Lazio region helps to demonstrate the dynamics of virus circulation in Italy. In particular, a small number of mutations have been detected in these strains, but the real impact and role that these mutations may have on the pathogenicity and transmissibility of SARS-CoV-2 remains to be determined.
A limitation of our research is that only a portion of viral sequences, including the sequences from Italy, have been published as of April 10, 2020; phylogenetic analysis could substantially change when more sequences are made available. Continued genomic surveillance strategies are needed to improve monitoring and understanding of current SARS-CoV-2 epidemics, which might help to lessen the public health impact of COVID-19. Furthermore, increased sequencing capacity is necessary for contact tracing and enhanced surveillance activity.
Dr. Bartolini is a senior scientist at Microbiology Laboratory and Infectious Diseases Biorepository at the National Institute for Infectious Diseases “L. Spallanzani.” Her primary research interests are next-generation sequencing and emerging and reemerging infections.
Acknowledgments
We thank the contributors of genome sequences of the newly emerging coronavirus (the originating and submitting laboratories) for sharing their sequences and other metadata through the GISAID Initiative, on which this research is based. We thank Salvatore Conti and Alessandro Albiero for their support in NGS sequencing and analysis.
The INMI sequences have been deposited in GISAID with accession IDs as follows: INMI3: EPI_ISL_ 417921; INMI4: EPI_ISL_417922; INMI5: EPI_ISL_ 417923; INMI6: EPI_ISL_ 419254; INMI7: EPI_ISL_ 419255; INMI 8: EPI_ISL_424342; INMI 9: EPI_ISL_424343; INMI 10: EPI_ISL_424344.
This research was supported by funds to National Institute for Infectious Diseases “Lazzaro Spallanzani” IRCCS from Ministero della Salute, Ricerca Corrente, linea1; European Commission–Horizon 2020 (EU project 653316-EVAg; EU project no. 101003544–CoNVat; EU project no. 101003551–EXSCALATE4CoV).
B.B. coordinated the experiments and wrote the manuscript; B.B. and M.R. performed the NGS experiment; G.M., F.C., F.Ca., E.L., L.B., C.C. performed SARS-CoV-2 diagnosis; F.V. performed the epidemiological analysis; C.E.M.G., F.M. and E.G. performed bioinformatic and phylogenetic analysis; M.R.C. and A.D.C. supervised the study design; G.I. read and revised the manuscript. All the authors read and approved the manuscript.
References
- World Health Organization (WHO). Coronavirus disease 2019 (COVID-19) situation report—81. 2020 Apr 10 [cited 2020 May 14]. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200410-sitrep-81-covid-19.pdf?sfvrsn=ca96eb84_2
- European Centre for Disease Control and Prevention (ECDC). COVID-19 situation update worldwide, as of 10 April 2020. 2020 Apr 10 [cited 2020 May 14]. https://www.ecdc.europa.eu/en/geographical-distribution-2019-ncov-cases
- Giovanetti M, Angeletti S, Benvenuto D, Ciccozzi M. A doubt of multiple introduction of SARS-CoV-2 in Italy: A preliminary overview. J Med Virol. 2020;1–3. DOIPubMedGoogle Scholar
- Stefanelli P, Faggioni G, Lo Presti A, Fiore S, Marchi A, Benedetti E, et al.; On Behalf Of Iss Covid-Study Group. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: additional clues on multiple introductions and further circulation in Europe. Euro Surveill. 2020;25. DOIPubMedGoogle Scholar
- Zehender G, Lai A, Bergna A, Meroni L, Riva A, Balotta C, et al. Genomic characterization and phylogenetic analysis of SARS-COV-2 in Italy. J Med Virol. 2020;1–4.PubMedGoogle Scholar
- Capobianchi MR, Rueca M, Messina F, Giombini E, Carletti F, Colavita F, et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin Microbiol Infect. 2020 Mar 27 [Epub ahead of print].
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleOriginal Publication Date: May 27, 2020
Table of Contents – Volume 26, Number 8—August 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Antonino Di Caro, National Institute for Infectious Diseases “Lazzaro Spallanzani,” IRCCS Via Portuense 292, 00149 Rome, Italy
Top