Volume 27, Number 1—January 2021
Research
Comparative Omics Analysis of Historic and Recent Isolates of Bordetella pertussis and Effects of Genome Rearrangements on Evolution
Ana Dienstbier, Fabian Amman, Denisa Petráčková, Daniel Štipl, Jan Čapek, Jana Zavadilová, Kateřina Fabiánová, Jakub Držmíšek, Dilip Kumar, Mark Wildung, Derek Pouchnik, and Branislav Večerek
Figure 1
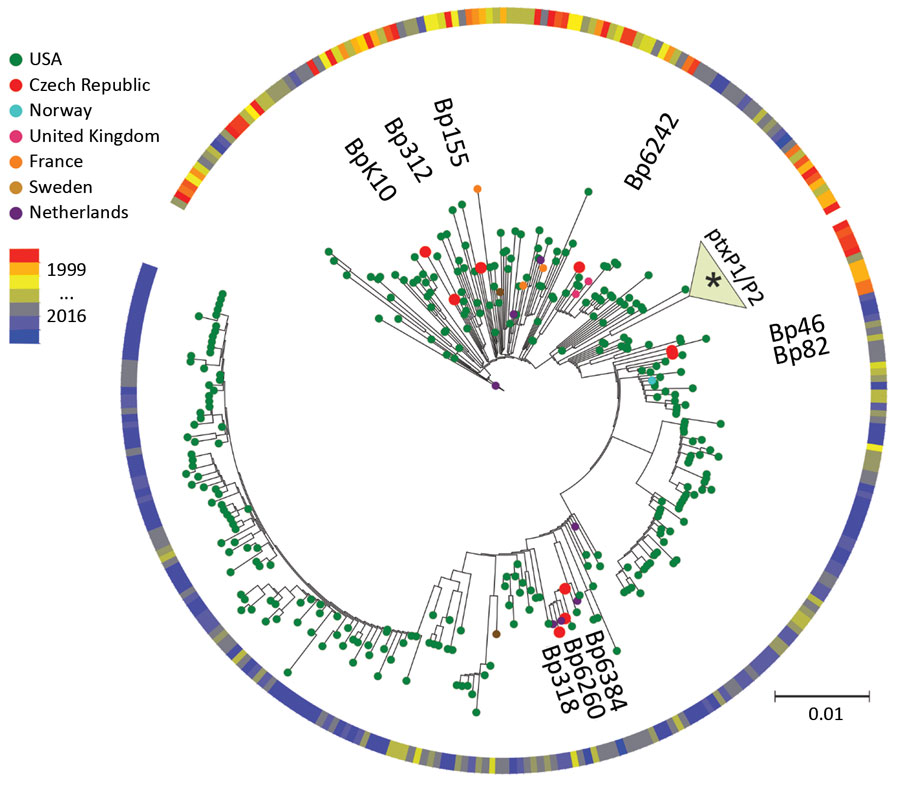
Figure 1. Maximum-parsimony, unrooted phylogenetic tree based on single-nucleotide polymorphism analysis of available genome sequences of Bordetella pertussis. Red dots indicate recent isolates from the Czech Republic. Year and country of isolation are color-coded. The 3 black dots indicate time span between 1999 and 2016. Asterisk (*) indicates association of historic strains from the Czech Republic with the ptxP1/ptxP2 clade. Scale bar indicates nucleotide substitutions per site.
Page created: September 01, 2020
Page updated: December 21, 2020
Page reviewed: December 21, 2020
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.