Volume 31, Number 5—May 2025
Dispatch
Rapid Transmission and Divergence of Vancomycin-Resistant Enterococcus faecium Sequence Type 80, China
Cite This Article
Citation for Media
Abstract
We investigated genomic evolution of vancomycin-resistant Enterococcus faecium (VREF) during an outbreak in Shenzhen, China. Whole-genome sequencing revealed 2 sequence type 80 VREF subpopulations diverging through insertion sequence–mediated recombination. One subpopulation acquired more antimicrobial resistance and carbohydrate metabolism genes. Persistent VREF transmission underscores the need for genomic surveillance to curb spread.
Vancomycin-resistant Enterococcus faecium (VREF) causes hospital-acquired infections worldwide and poses a threat to public health (1). Whole-genome sequencing (WGS) has demonstrated that new healthcare-associated E. faecium clones rapidly emerge, evolve, and adapt through intragenus recombination, displacing existing clones (2,3).
During the past decade, clonal complex 17 sequence type (ST) 80 rapidly became the predominant VREF lineage in Denmark (4), Australia (5), Ireland (6), Spain (7), and Sweden (8) and accounted for 40%–67.1% of VREF isolates disseminated in hospital settings. Few ST80 cases were reported in Asia countries, including China, until an independent lineage of ST80, sequence cluster (SC)11, emerged in January 2021 as the predominant cause of an ongoing VREF outbreak in Guangdong Province (9) and posed a risk of spreading to other areas. However, the pangenomic features and adaptation potential of the emerging SC11 remain unknown.
Figure 1

Figure 1. Evolution and variation of SC11 inferred from core genome SNPs during rapid transmission and divergence of vancomycin-resistant Enterococcus faeciumST80, China. Graphs shows reconstructed tree from core genome SNPs...
VREF isolation rates also substantially increased in the metropolitan city of Shenzhen, Guangdong Province, China, during 2021–2024. VREF isolation rates before 2021 remained <6% (predominantly <5%) but rose to an average of 11.53% in 2024 (Appendix 1 Figure 1, panel A). To trace the source and characterize genomic features that potentially contributed to outbreaks, we conducted a multicenter genomic epidemiology study and integrated pangenomic variation analysis.
We performed WGS analysis (Appendix 1) on 42 VREF isolates (primarily collected after April 2022) from 39 patients across 7 hospitals, designated SZ_A through SZ_F, including 2 affiliated hospitals, SZ_C1 and SZ_C2, grouped as SZ_C. We used WGS to identify STs and used phylogenetic analysis to determine ST sources in a global context. We assessed genetic diversity, indicating mutation rates during circulation, using pairwise core genome single-nucleotide polymorphism (cgSNP) distance. We characterized population structure to show divergence and emergence of novel variant populations.
Among the 42 isolates, 34 (81%) were ST80 isolates, 7 (17%) were ST80 variant isolates (ST80_GDvariant1) with ddl loci mutated from 1 to 194, and 1 (2%) was an ST78 isolate that was collected in 2023 (Appendix 1 Figure 1, panel B; Appendix 2). Unexpectedly, ST80_GDvariant1 isolates independently emerged in 5 branches and did not originate from a single mutation event (Figure 1; Appendix 1 Figure 2, panel A).
Phylogenetic analysis inferred from cgSNP of the 41 isolates (ST80 and variants) and 703 other publicly available ST80 isolates revealed that the 41 isolates from the ongoing outbreak in Guangdong are affiliated with SC11 (Appendix 1 Table 2, Figure 2, panel A) (9). Within SC11, two 2022 strains from Guangzhou, SZYSC_ZDYVRE007 and SZYSC_ZDYVRE008G, with a 5-SNP divergence, clustered adjacent to the lineage root (Figure 1; Appendix 1 Figure 2, panel A) and formed a distinct VREF sublineage (SC11-root sublineage) with a 37-SNP average divergence from other SC11 strains (bootstrap = 72) (Figure 1). In contrast, the remaining SC11 strains exhibited tight clustering (pairwise distances of <5 SNPs [36.5%] and 5–10 SNPs [49.8%]) and formed a dominant clade (SC11-outbreak sublineage) (Figure 1). Those findings suggest that the SC11-outbreak lineage originated from a single progenitor or highly related lineage, enabling rapid transmission.
Figure 2
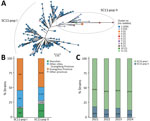
Figure 2. Lineages detected in a study of rapid transmission and divergence of vancomycin-resistant Enterococcus faeciumST80, China, showing 2 lineages circulating in parallel. A) SC11 subpopulations labeled on pangenomic tree...
To resolve strain differentiation, we analyzed SC11-outbreak_lineage population structure by using PopPUNK (https://github.com/bacpop/PopPUNK), which integrates core and accessory genomic variation (10). We delineated 2 subpopulations, SC11-pop I and SC11-pop II (Figure 2, panel A; Appendix 1 Figure 4). SC11-pop II isolates formed tight clusters in the pangenome-based tree (Figure 2, panel A) but dispersed in the cgSNP phylogeny (Figure 1), suggesting subpopulation divergence was primarily driven by gene gain or loss.
Figure 3
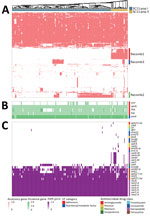
Figure 3. Alignment of SC11 to determine divergence during rapid transmission and divergence of vancomycin-resistant Enterococcus faeciumST80, China. To determine the genes associated with the divergence of SC11 we aligned...
SC11-pop I and SC11-pop II circulated in parallel for >3 years (2021–2024), and SC11-pop II showed broader transmission (Figure 2, panel B) and maintained ≈15% prevalence (Figure 2, panel C). SC11-pop II showed higher prevalence than SC11-pop I in Shenzhen and other provinces (Figure 2, panel B). Genetically, SC11-pop II exhibited enhanced horizontal gene transfer activity, carrying more insertion sequences (ISs) (Appendix 1 Figure 5), plasmid-like elements (Appendix 1 Figure 6), and diverse antimicrobial resistance genes (Figure 3). Although all SC11 isolates harbored the VanA operon, 8 SC11-pop II strains uniquely acquired VanM operon (Figure 3). SC11-pop II cases showed trends of increased hospitalization and underlying conditions, including hypertension and cardiovascular, respiratory, and kidney diseases, but statistical significance was limited by the sample size (Appendix 1 Table 5). Expanded surveillance is required to clarify clinical distinctions between SC11-pop I and SC11-pop II.
To identify divergence drivers, we compared core genomic mutations and accessory gene variations between SC11-pop I and SC11-pop II. Unexpectedly, no substantial cgSNP differences emerged (Appendix 3), indicating divergence was not driven by core genome mutations. The SC11 lineage pangenome (n = 235) comprised 3,674 genes, including 2,367 core genes and 1,307 accessory genes, an accessory-to-core gene ratio of 0.55. Concerning gene gain or loss, SC11-pop II specifically acquired 152 genes (forming Recomb1 modular) and showed higher frequency of 13 other functional unknown genes (except endoribonuclease PemK) and lower frequency of 9 genes versus SC11-pop I (p<0.05) (Appendix 1 Table 6), indicating greater exogenous gene acquisition. As was observed in the hierarchical clustering of accessory genes on the pangenomic phylogeny, we identified 3 recombination gene modules (Recomb1–3) that were frequently acquired in SC11 (Figure 3, panel A). Recomb1 was exclusive to SC11-pop II (31/35 strains; 1 strain carries more than half of Recomb1 genes and is recognized as Recomb1 positive, similar to the standard for recognizing positivity in Recomb2 and Recomb3), whereas Recomb2 (68 strains) and Recomb3 (23 strains) occurred in both populations (Figure 3, panel A). The fixation of Recomb1–3 suggested their roles in emergence and adaptation of novel variants in SC11. Of note, Recomb1 contained 11 carbohydrate metabolism genes (Appendix 1 Table 4), which are known factors in E. faecium that contribute to clinical adaption and epidemics of E. faecium (12). The stable 3-year persistence of Recomb1 in SC11-pop II across regions suggests a functional importance in host adaptation and potential virulence.
Recombination surpasses mutation as the primary driver of E. faecium genetic diversity (11), and IS-mediated events occur within days during infection (13). Ten families of IS elements were found in all SC11 isolates (Appendix 1 Figure 5). Enhanced IS transposition was associated with rapid core gene mutation (Appendix 1). Recomb1–3 acquisitions were linked to IS-mediated recombination, primarily involving genes related to DNA transposition, replication, or recombination (Appendix 1 Table 4). Recomb1 contained more ISs than Recomb2 or Recomb3, and IS91 was exclusively acquired by SC11-pop II. High-frequency modular recombination in Recomb1 involved IS91, ISL3, and IS256 (specific to SC11-pop II), whereas IS200/IS605 and IS1182 occurred at lower frequencies (Table; Appendix 1 Figure 5). Recomb2 in SC11-pop I occasionally incorporated IS3 alongside ISL3, IS66, IS982, IS256, or IS30 (Table). Recomb3 exclusively associated with IS3 in all 7 SC11-pop II isolates but was absent in SC11-pop I (Table). No plasmid marker genes co-occurred with Recomb1–3, except 9 Recomb1 genes colocalized with MOBT (plasmid relaxase) on contig AXARS010000069.1 (strain SZYSC_22VRE31), suggesting that plasmids did not directly transmit Recomb1–3.
Identifying SC11’s most recent ancestor is crucial for elucidating its evolutionary mechanism and mitigating emergent threats. We hypothesize a shared ancestry between SC11-root and SC11-outbreak sublineages. Expanded surveillance of outbreak-associated hospitals and retrospective analysis of pre-2021 VREF isolates are needed to trace the origin.
In summary, we showed that increasing VREF prevalence in Shenzhen, China, constitutes part of the ongoing SC11 outbreak, likely originating from Guangzhou. Population structure analysis revealed 2 stable, circulating SC11 subpopulations, emergence of which was driven by IS-mediated recombination. Sustained surveillance of those subpopulations is essential to prevent the emergence of high-risk clones with increased transmissibility and virulence.
Dr. Li is a research scientist at the National Clinical Research Center for Infectious Diseases, Shenzhen, China. His research interests include genomic epidemiology and the mechanisms underlying epidemic spread of pathogens.
Acknowledgments
The whole-genome assemblies produced in this study are deposited in GenBank (BioProject accession no. PRJNA1221312).
This study was approved by the Bioethics Committee of Shenzhen Third People’s Hospital (ref. no. 2024-009-02). All methods were performed in accordance with the relevant guidelines and regulations.
This work was funded by the Guangdong Basic and Applied Basic Research Foundation (grant no. 2024A1515010319), the Science and Technology Program of Shenzhen (grant nos. KCXFZ20230731100901003 and KJZD20230923115116032), the Shenzhen Key Laboratory of Biochip (grant no. ZDSYS201504301534057), and the Shenzhen High-Level Hospital Construction Fund.
Author contributions: J.Q. and L.L. were involved in the conceptualization. Y.X., B.F., J.W., J.Z., Z.L., Y.Z., H.M., L.Z., D.L., D.G., S.Y., and L.L. were involved in laboratory experiments and data collection. L.L., X.W., and Y.W. were involved in data curation. X.W., L.L., and Y.W. were involved in data analysis and results visualization. J.Q. acquired the funding acquisition. L.L. and X.W. were involved in writing the original draft. J.Q. and L.L. were involved in revising and editing the manuscript. All authors contributed to the article and approved the submitted version.
References
- Van Tyne D, Gilmore MS. Friend turned foe: evolution of enterococcal virulence and antibiotic resistance. Annu Rev Microbiol. 2014;68:337–56. DOIPubMedGoogle Scholar
- Gao W, Howden BP, Stinear TP. Evolution of virulence in Enterococcus faecium, a hospital-adapted opportunistic pathogen. Curr Opin Microbiol. 2018;41:76–82. DOIPubMedGoogle Scholar
- Gorrie C, Higgs C, Carter G, Stinear TP, Howden B. Genomics of vancomycin-resistant Enterococcus faecium. Microb Genom. 2019;5:
e000283 . DOIPubMedGoogle Scholar - Pinholt M, Bayliss SC, Gumpert H, Worning P, Jensen VVS, Pedersen M, et al. WGS of 1058 Enterococcus faecium from Copenhagen, Denmark, reveals rapid clonal expansion of vancomycin-resistant clone ST80 combined with widespread dissemination of a vanA-containing plasmid and acquisition of a heterogeneous accessory genome. J Antimicrob Chemother. 2019;74:1776–85. DOIPubMedGoogle Scholar
- Pratama R, Beukers AG, McIver CJ, Keighley CL, Taylor PC, van Hal SJ. A vanA vancomycin-resistant Enterococcus faecium ST80 outbreak resulting from a single importation event. J Antimicrob Chemother. 2021;77:31–7. DOIPubMedGoogle Scholar
- Egan SA, Kavanagh NL, Shore AC, Mollerup S, Samaniego Castruita JA, O’Connell B, et al. Genomic analysis of 600 vancomycin-resistant Enterococcus faecium reveals a high prevalence of ST80 and spread of similar vanA regions via IS1216E and plasmid transfer in diverse genetic lineages in Ireland. J Antimicrob Chemother. 2022;77:320–30. DOIPubMedGoogle Scholar
- Rodríguez-Lucas C, Fernández J, Raya C, Bahamonde A, Quiroga A, Muñoz R, et al. Establishment and persistence of glycopeptide-resistant Enterococcus faecium ST80 and ST117 clones within a health care facility located in a low-prevalence geographical region. Microb Drug Resist. 2022;28:217–21. DOIPubMedGoogle Scholar
- Fang H, Fröding I, Ullberg M, Giske CG. Genomic analysis revealed distinct transmission clusters of vancomycin-resistant Enterococcus faecium ST80 in Stockholm, Sweden. [</jrn>]. J Hosp Infect. 2021;107:12–5. DOIPubMedGoogle Scholar
- Shen C, Luo L, Zhou H, Xiao Y, Zeng J, Zhang L, et al. Emergence and ongoing outbreak of ST80 vancomycin-resistant Enterococcus faecium in Guangdong province, China from 2021 to 2023: a multicenter, time-series and genomic epidemiological study. Emerg Microbes Infect. 2024;13:
2361030 . DOIPubMedGoogle Scholar - Lees JA, Harris SR, Tonkin-Hill G, Gladstone RA, Lo SW, Weiser JN, et al. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 2019;29:304–16. DOIPubMedGoogle Scholar
- Palmer KL, Schaik WV, Willems RJL, Gilmore MS. Enterococcal genomics. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: from commensals to leading causes of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary; 2014. 193–230.
- Chilambi GS, Nordstrom HR, Evans DR, Ferrolino JA, Hayden RT, Marón GM, et al. Evolution of vancomycin-resistant Enterococcus faecium during colonization and infection in immunocompromised pediatric patients. Proc Natl Acad Sci U S A. 2020;117:11703–14. DOIPubMedGoogle Scholar
- Udaondo Z, Abram KZ, Kothari A, Jun SR. Insertion sequences and other mobile elements associated with antibiotic resistance genes in Enterococcus isolates from an inpatient with prolonged bacteraemia. Microb Genom. 2022;8:
mgen000855 . DOIPubMedGoogle Scholar - Xie Z, Tang H. ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics. 2017;33:3340–7. DOIPubMedGoogle Scholar
Figures
Table
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 31, Number 5—May 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jiuxin Qu, Department of Clinical Laboratory, Shenzhen Third People’s Hospital, National Clinical Research Center for Infectious Diseases, Second Affiliated Hospital of Southern University of Science and Technology, No. 29 Bulan Rd, Longgang District, Shenzhen 518000, China
Top