Volume 31, Number 5—May 2025
Research
Metagenomic Identification of Fusarium solani Strain as Cause of US Fungal Meningitis Outbreak Associated with Surgical Procedures in Mexico, 2023
Cite This Article
Citation for Media
Abstract
We used metagenomic next-generation sequencing (mNGS) to investigate an outbreak of Fusarium solani meningitis in US patients who had surgical procedures under spinal anesthesia in Matamoros, Mexico, during 2023. Using a novel method called metaMELT (metagenomic multiple extended locus typing), we performed phylogenetic analysis of concatenated mNGS reads from 4 patients (P1–P4) in parallel with reads from 28 fungal reference genomes. Fungal strains from the 4 patients were most closely related to each other and to 2 cultured isolates from P1 and an additional case (P5), suggesting that all cases arose from a point source exposure. Our findings support epidemiologic data implicating a contaminated drug or device used for epidural anesthesia as the likely cause of the outbreak. In addition, our findings show that the benefits of mNGS extend beyond diagnosis of infections to public health outbreak investigation.
Whole-genome sequencing (WGS) of pathogens, including viruses, bacteria, and fungi, is a vital tool for detecting and investigating outbreaks (1–3). In addition to providing definitive identification of the causative organism, WGS can be used to construct phylogenetic trees, which in turn can be used to detect more cases, resolve disease transmission patterns, and identify potential outbreak sources. However, conventional WGS depends on the ability to grow the pathogen in culture. For fastidious organisms such as mycobacteria, fungi, and some viruses, successful culturing is often difficult and slow; growth of the organism requires weeks to months and sometimes is not possible at all. To bypass the requirement for culture, techniques such as multilocus sequence typing/multilocus sequence analysis (MLST/MLSA) have been developed, whereby specific regions of the microbial genome (loci) are targeted for PCR amplification directly from primary clinical material (4,5). A key limitation of MLST/MLSA, however, is the need to have the target organism identified, loci defined, and primers at hand a priori. Thus, MLST/MLSA is typically not useful for immediate outbreak situations caused by rare or unexpected pathogens.
Metagenomic next-generation sequencing (mNGS) is an agnostic diagnostic method with the potential to identify any pathogen, whether bacterial, viral, parasitic, or fungal, on the basis of shotgun sequencing of DNA or RNA (6). The use of mNGS testing from clinical samples has been shown to increase diagnostic yield and provide actionable results in multiple prospective studies (7–11). In addition, mNGS has proven to be an invaluable tool for identifying and initially characterizing emerging pathogens, such as SARS-CoV-2 (12). However, the use of mNGS to date has been mainly focused on pathogen detection for clinical diagnostic purposes, rather than outbreak investigation, because coverage of the pathogen genome recovered by mNGS is generally sparse and uneven.
On May 8, 2023, the Centers for Disease Control and Prevention (CDC) Emerging Infections Network reported cases of suspected fungal meningitis in US patients who had undergone surgical procedures performed under epidural anesthesia in the city of Matamoros in Durango state, Mexico (13). Using clinical mNGS testing, the University of California, San Francisco (UCSF), clinical microbiology laboratory reported identification of Fusarium solani species complex in cerebrospinal fluid from an affected patient (P1) with probable fungal meningitis in the United States on May 28, 2023 (14). That initial case and additional cases were subsequently confirmed independently by panfungal PCR followed by sequencing of the amplicon to confirm detection of F. solani (13). During the outbreak, a total of 184 patients in 22 US states were identified as persons potentially exposed, among whom 24 were identified with fungal meningitis and 12 died, mainly from severe vascular complications (13,15). Here, we describe a novel analytic technique called metaMELT, or metagenomic multiple extended locus typing, as a tool for simultaneously diagnosing infections and characterizing the interrelatedness of F. solani strains in patients from the Matamoros outbreak.
Human Sample Collection and Processing
Cerebrospinal fluid (CSF) samples from 5 patients (P1–P5) in the Matamoros outbreak were available for analysis. A CSF sample from patient P1 was sent to the UCSF Clinical Microbiology Laboratory for CSF mNGS testing and was processed and sequenced as previously described (16). Residual CSF, plasma, and brain tissue biopsy samples from patients P1–P4 were also processed and sequenced using mNGS. Cultures of F. solani were also obtained from P1 and P5 CSF samples.
For P1–P4 samples, we extracted DNA by using the MagMAX Viral/Pathogen II (MVP II) Nucleic Acid Isolation Kit (Thermo Fisher Scientific, https://www.thermofisher.com) and the KingFisher Flex Purification System with a 96 deep-well head (Thermo Fisher Scientific). We loaded the extracted DNA on the MagicPrep NGS instrument (Tecan Genomics, Inc., https://www.tecan.com) to undergo end repair, adaptor ligation and barcoding, amplification (25 cycles), and purification. We enriched some of the libraries for microbial DNA by using DepleteX (Jumpcode Genomics, https://www.jumpcodegenomics.com), an early release of a CRISPR-based human DNA depletion kit, according to the manufacturer’s specifications. That kit leverages Cas9 depletion and exonuclease activity to efficiently remove human DNA from samples with high human content.
CRISPR-based host depletion increased the number of Fusarium-specific reads in the P1 CSF DNA library from 13 to 223 (16.4-fold enrichment increase), corresponding to an increase of 1.5 to 2.4 reads per million (RPM), a 1.6-fold enrichment increase. In the P3 plasma DNA library, the number of reads decreased from 60 to 36 after enrichment, but that decrease corresponded to an RPM increase from 0.12 to 0.16 (1.3-fold enrichment increase). We quantified and normalized the libraries by using the Qubit dsDNA HS Assay on the Qubit Flex (Thermo Fisher Scientific). We sequenced final pooled libraries as single-end reads on the NextSeq 550 (Illumina, https://www.illumina.com) by using the High-Output Kit or the NovaSeq Kit (Illumina) at 150 cycles.
F. solani WGS
We performed WGS of 18 F. solani isolates, including isolates cultured from 2 patients (P1 and P5) in the Matamoros outbreak as follows. We extracted DNA by using the DNeasy Blood and Tissue kit (QIAGEN, https://www.qiagen.com), and then used the NEBNext Ultra DNA Library Prep kit (New England Biolabs, https://www.neb.com) to construct DNA fungal genomic libraries for Illumina sequencing. We sequenced the isolate from patient P1 (genome B27264) on the Illumina MiSeq (250-bp paired-end sequencing, or 500 cycles). We sequenced the isolate from patient P5 (genome B27166) and 16 additional F. solani genomes unrelated to the Matamoros outbreak by using the NovaSeq 6000SP Reagent Kit (Illumina) at 500 cycles.
Bioinformatic Methods
We used the SURPI+ computational pipeline version 1.0.0 (https://github.com/chiulab/SURPI-plus-dist), run as a container on either a secure server or cloud infrastructure, to identify pathogens from mNGS data (16,17). We preprocessed reads by trimming adapters and removing low-complexity and low-quality sequences and then performed computational subtraction of human reads by using SNAP version 1.0 (16) and Bowtie2 version 2.3.2 GTCh38/hg38 build (https://sourceforge.net/projects/bowtie-bio) to align and exclude reads mapping to the human genome. We aligned the remaining nonhuman reads to microbial reference sequences in the National Center for Biotechnology Information (NCBI) nucleotide database (https://www.ncbi.nlm.nih.gov/nucleotide; March 2019 build) using the SNAP nucleotide aligner (18). We performed filtering and taxonomic classification algorithms to remove spurious hits and classify each read at the species, genus, or family level, as previously described (16).
We further analyzed CSF mNGS reads from P1 by nucleotide BLASTn (https://blast.ncbi.nlm.nih.gov) alignment to the genome of an F. solani reference strain (ATCC strain no. MYA-4622) with an E-value of 1 × 10−8 and the remaining parameters set to corresponding default values (19). We visualized mapped reads by using Ensembl Fungi (20). Among the 28 F. solani cultured isolates included in the analysis, 10 were from genomes downloaded from GenBank. For the other 18 newly sequenced isolates, we obtained draft genomes by de novo assembly of raw reads using the SPADES genome assembler version 3.15.4 at default parameters (21).
We wrote Linux scripts and code to perform the metaMELT method for each patient as follows. For reads from patients P1–P4 identified as F. solani in SURPI+, we trimmed to the desired length and successively aligned reads by using BLASTn at a 1 × 10−8 E-value cutoff for each of the 28 F. solani reference genomes, including the assembled genomes from P1 and P5. If an alignment was successful, we extracted the mapped region corresponding to the read from the reference genome; otherwise, the system generated a synthetic dummy read consisting of a stretch of Ns (ambiguous nucleotides). We then concatenated reads from all 4 patients and the corresponding mapped regions in each reference genome by using a 50-bp spacer of Ns to separate each read or region. We performed multiple sequence alignment of the concatenated sequences by using MAFFT version 7.388 (22) and the following parameters: FFT-NS-2 algorithm, 200 PAM/k = 2 scoring matrix, gap open penalty = 3, and offset value = 0.123. We then constructed phylogenetic trees by using PhyML 3.0 (23) and substitution model TN93 with SH-like branch supports, in Geneious version 11.1.5 (24).
Inclusion and Ethics
Sequencing data from clinical CSF mNGS testing and deidentified residual samples from hospitalized patients who were part of the F. solani outbreak were analyzed under protocols approved by the UCSF institutional review board (protocol no. 11-05519). Clinical and demographic patient-level data were not collected because that information was not considered relevant to this study. All patients with sufficient remaining volume of residual samples were included in the study.
Figure 1

Figure 1. Identification of Fusarium solaniin CSF from a patient with fungal meningitis associated with surgical procedures in Matamoros, Mexico, 2023. A) Heat map of 13 mNGS CSF reads from...
We used clinical CSF mNGS testing and SURPI+ analysis to diagnose F. solani infection in case P1. Among the 8.7 million reads that we recovered from CSF, 13 (0.00015%) aligned most closely to the genome of Nectria hematococca, the anamorph of F. solani (Figure 1, panel A). We did not observe any co-infections from viruses, bacteria, or parasites. The default SURPI+ pipeline uses the GenBank nucleotide database for its microbial reference database (16,17), which does not contain any fungal whole-genome sequences. Because fungal whole-genome sequences are found in the GenBank whole-genome shotgun database (25), we used that database to align the mNGS reads directly to an F. solani reference genome (ATCC strain no. MYA-4622) and identified an additional 5 fungal reads. We found that the 18 total mapped F. solani reads were distributed randomly across the ≈53 Mb genome (Figure 1, panel B).
We subsequently obtained more F. solani reads from the P1 CSF sample by additional sequencing and we also sequenced CSF, plasma, or brain tissue from 3 more patients (P2, P3, and P4) in the Matamoros outbreak who had PCR-confirmed F. solani meningitis. To maximize recovery of F. solani reads, we sequenced mNGS libraries both without and with enrichment for microbial DNA using DepleteX, although the level of enrichment was only 1.3–1.6 RPM using that kit. The total number of F. solani reads recovered from each patient ranged from 263 to 187,773.
Figure 2
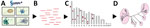
Figure 2. Flowchart showing the metaMELT analysis workflow used for identification of Fusarium solanistrain as cause of fungal meningitis US outbreak associated with surgical procedures in Mexico, 2023. metaMELT (metagenomic...
Figure 3

Figure 3. Diagram of concatenation and alignment step of the metaMELT procedure (metagenomic multiple extended locus typing, a novel analytic technique for simultaneously diagnosing the infection and characterizing the interrelatedness of ...
The number of fungal mNGS reads was not sufficient to assemble the F. solani genome from any single patient. To achieve 30× coverage, we estimated that ≈10.6 million 150-bp F. solani–specific reads would need to be sequenced. The actual read numbers and percent coverage achieved from CSF mNGS from infected patients was extremely low; the number of reads ranged from 263 to 187,773 and the coverage ranged from 0.0005% to 0.35% of the fungal genome (Table 1). Thus, we developed metaMELT as a tool to enable comparison of fungal strains from individual patients using sparse mNGS data. The metaMELT method involves first extracting the regions defined by randomly selected mNGS reads from each patient sample from their corresponding locations (loci) in all available F. solani reference genomes (n = 28) (Figure 2). The total number of extracted reads or loci is equal to the product of the number of samples and the number of mNGS reads that are randomly selected from each sample. After concatenating the extracted reads, we performed phylogenetic analysis of the concatenated sequences, with each sequence derived from a single patient or a reference genome (Figure 3).
Figure 4
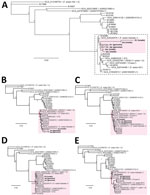
Figure 4. Phylogenetic analysis of concatenated metagenomic next-generation sequencing reads from US patients from a fungal meningitis outbreak associated with surgical procedures in Mexico, 2023. A) Phylogenetic trees showing clustering of strains...
By early 2024, only 10 complete F. solani reference genomes were available for download in the GenBank whole-genome shotgun database (Figure 4, panel A). We performed de novo assembly on additional draft genomes from 18 cultured F. solani isolates in the CDC fungal biorepository from raw next-generation sequence data and included those in the phylogenetic analysis (Figure 4, panel B). Each draft genome consisted of a series of contigs (26), ranging from 1,057 to 7,216 contigs (Table 2). Of note, 2 draft genomes, B27264 from P1 and B27166 from P5, had been newly assembled from cultured outbreak isolates; patient P5 was part of the Matamoros outbreak for whom residual CSF was not available for mNGS. The accuracy of phylogenetic analysis relied on polymorphisms between an outbreak strain or cultured isolate and each reference genome in the database and not on polymorphisms between individual strains or isolates, because the sparse mNGS reads recovered from the ≈53 Mb genome were unlikely to overlap.
We performed phylogenetic analysis of the concatenated sequences from P1−P4 and the 28 reference genomes. To evaluate the flexibility of metaMELT for different sample types, P1 and P2 included CSF reads, P3 included only plasma reads (n = 96), and P4 included mostly brain biopsy tissue reads (608 of 612 reads). Each concatenated sequence consisted of 90 randomly selected mNGS reads of 150-bp length because only 96 total plasma mNGS reads were available for patient P3. The topology of the resulting tree revealed that the F. solani strains from all 5 patients in the Matamoros outbreak were positioned together in a single cluster (Figure 4, panel A). To account for potential bias in the phylogenetic estimates because of the high proportion of ambiguous nucleotides in each of the concatenated sequences from patients P1–P4 (27), we reran the phylogenetic analysis including only 1 patient at a time, and all trees positioned the individual patient in the same cluster (Figure 4, panels B–E).
Figure 5
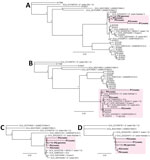
Figure 5. Effect of number of reference genomes on performance of metaMELT (metagenomic multiple extended locus typing, a novel analytic technique for simultaneously diagnosing the infection and characterizing the interrelatedness of ...
Next, we assessed whether clustering of mNGS reads from patients P1–P4 could still be visualized if only 1 outbreak-related reference genome from P5 was available for comparison or if no outbreak-related reference genome was available. We observed clustering of patients P1–P4 for both analyses (Figure 5, panels A, B), albeit with 11 unrelated genomes assigned to the cluster if no outbreak-related genome was included (Figure 5, panel B). Because only a few reference genomes might be in the existing reference databases for a given target pathogen, we then determined how the total number of available reference genomes affected patient clustering by metaMELT. Despite inclusion of only 10 or 5 total reference genomes, we still observed clustering of patients P1–P5 by phylogenetic analysis (Figure 5, panels C, D).
Based on MLST/MLSA analysis of the concatenated ITS, rpb2, and tef1 genes (28), the predicted nucleotide pairwise identity between the 2 draft genomes from the Matamoros outbreak, B27264 (P1) and B27166 (P5), was 96% (Appendix Figure 1, panel A), and overall pairwise identities for the 28 reference genomes ranged from 89% to 96%. The high pairwise identity between the P1 and P5 isolates and positioning in the same subcluster by phylogenetic analysis (Appendix Figure 1, panel B) supported the notion of a single F. solani strain as the cause of the outbreak. Of note, the larger cluster consisting of an additional 11 genomes unrelated to the outbreak (Appendix Figure 1) was the same cluster that included the concatenated mNGS sequences from cases P1–P4 in all previous metaMELT phylogenetic analyses (Figures 4, 5).
We sought to ascertain the effect of read lengths and numbers on the clustering of the F. solani strains from patients P1−P5 by metaMELT. All 4 sequenced strains were correctly clustered at read lengths of >100 bp and >40 mNGS reads (Appendix Figure 2, panel A). To establish a quality control criterion for metaMELT at the empirically determined 40-read cutoff (Appendix Figure 2, panel A), we performed 20 bootstrap replicates in which we randomly sampled and analyzed 40 100-bp reads from patients P1–P4 by metaMELT. Phylogenetic trees from all 20 replicates correctly positioned patients P1–P4 in the same cluster (Appendix Figure 2, panel B).
In this study, we used clinical mNGS testing to identify F. solani in a patient from a fungal meningitis outbreak associated with surgical procedures in Matamoros, Mexico. We used a novel analytic method, metaMELT, to leverage the mNGS data to aid the public health investigation. The metaMELT method is a powerful tool for species identification and evaluation of the interrelatedness of outbreak strains for pathogens that are difficult to culture. Clinical samples from 4 infected patients (P1–P4) and cultured isolates from P1 and another patient from the outbreak (P5) clustered together by phylogenetic analysis, showing that the strains from all 5 patients are the same or closely related. Those results indicate a high likelihood that all 5 infections, and, by extension, the US outbreak, originated from a single point source. The results of the CDC epidemiologic investigation, taken together with our findings, suggest a single point source and implicate a contaminated drug or device used for epidural anesthesia, either at the site of manufacturing or from breakdown of infection control practices at the clinics, as the likely cause of the outbreak.
The minimum number of reads necessary for accurate metaMELT analyses is dependent on multiple factors, including the sequence diversity of the species’ genome, availability of closely related reference genomes, and the quality and length of reads. Given the high levels of sequence diversity and large number of potential outbreak pathogens, we propose an empirically derived quality control criterion for investigation of interrelatedness of outbreak samples that uses bootstrap sampling of randomly selected mNGS reads and a >95% accuracy cutoff (i.e., at least 19 of 20 trees with the correct topology) (Appendix Figure 2, panel B).
The usefulness of metaMELT for analysis of the Matamoros outbreak described is likely due in part to the high sequence diversity of circulating Fusarium species isolates. In a previously published international outbreak of F. keratitis associated with contact lens wear, 19 of 39 isolates tested had a unique multilocus genotype (29). For other pathogens with lower genetic diversity, the usefulness of metaMELT may be limited because determining the relatedness of outbreak strains can be challenging. Another potential limitation of metaMELT is the need for enough complete reference genomes in publicly available databases for comparison, which might not be the case for rare or unusual pathogens.
Our study also extends the utility of mNGS and subsequent metaMELT analyses to not only agnostic diagnosis of infections in clinical settings but also the monitoring and tracking of communicable diseases, which are relevant to infection control, public health surveillance, and outbreak investigation. To date, those activities have largely relied on microbial WGS and phylogenetic analysis of the assembled sequences. However, WGS can be problematic because many atypical bacteria, fungi, and viruses grow slowly in culture or do not grow at all. The requirement for culture also inevitably delays the generation of actionable results in time-critical scenarios, such as outbreak investigation. Targeted methods, such as tiled multiplex amplicon sequencing for viruses (30) and MLST/MLSA for bacteria, parasites, and fungi (4,5,31,32), including F. solani (28,33), can be applied in lieu of WGS-based phylogenetic analysis. However, those approaches require that the pathogen be identified a priori and depend on the assays being immediately available at the time of identification, which is usually not the case with rare or unexpected pathogens such as F. solani. Clustering and phylogeny inferred from MLST/MLSA are also known to be inferior for some organisms compared to more detailed WGS analyses (34,35). Unlike WGS, metaMELT can be useful with mNGS reads that can vary by several orders of magnitude in number from sample to sample but typically produce very sparse coverage of the genome. In addition, metaMELT analysis enables the leveraging of mNGS data generated at the time of diagnosis and thus has the potential of providing immediate and actionable information to guide infection control and public health efforts.
In summary, our mNGS findings support epidemiologic data implicating a contaminated drug or device used for epidural anesthesia as a common point source and the likely cause of a F. solani meningitis outbreak in US patients associated with surgical procedures in Matamoros, Mexico. In addition, our findings show that mNGS could have benefits that extend beyond diagnosis of infections to more broadly assist in outbreak investigations.
Dr. Chiu is a professor in Laboratory Medicine and Infectious Diseases at University of California, San Francisco (UCSF), director of the UCSF Clinical Microbiology Laboratory, and a Chan-Zuckerberg Biohub Investigator. His research interests focus on clinical metagenomic assay development for infectious diseases and characterization of emerging outbreak viruses.
Author contributions: C.Y.C conceived and designed the study. V.S., N.S., A.F, L.G., L.P., and E.M. processed samples. C.Y.C., F.M.C., N.W.-B., P.R., L.N.S.H., D.C.G., L.S., S.W., J.K., J.A.W.G., L.G., L.P., I.G., and I.S.G. collected samples and clinical metadata. C.Y.C., V.S., M.D.L., P.B., E.M., D.J.S., and A.P.L. analyzed data. C.Y.C., P.R., J.K., T.M.C., S.V.B., D.J.S., A.P.L, and N.A.C. provided study oversight. C.Y.C. prepared the figures and wrote the manuscript. C.Y.C., M.D.L., P.B., P.R., H.R.V., E.M., D.J.S., A.P.L., and N.A.C. edited the manuscript. All authors read the manuscript and agree to its contents.
Acknowledgments
We thank the staff at the UCSF Clinical Microbiology Laboratory for help in processing patient CSF samples for clinical mNGS testing.
Raw FASTQ sequence metadata, including deidentified mNGS data from patients with Fusarium solani infection depleted of human genomic sequences, were submitted a Zenodo data repository (https://doi.org/10.5281/zenodo.12572958) and the Sequence Read Archive database (BioProject accession no. PRJNA1130911 and umbrella BioProject accession no. PRJNA171119). Custom scripts and code for running metaMELT and draft genome assemblies from 18 cultured isolates of F. solani have also been deposited into the Zenodo data repository. Further information and requests for resources and reagents should be directed to and will be fulfilled by Charles Y. Chiu (
This work was financially supported in part by funding from the following to C.Y.C.: BARDA EZ-BAA award no. 75A50122C00022, US Centers for Disease Control and Prevention grant nos. 75D30122C15360 and 75D30121C12641, Abbott Laboratories, and the Chan-Zuckerberg Biohub. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
C.Y.C. is a founder of Delve Bio and on the scientific advisory board for Delve Bio, Flightpath Biosciences, Biomeme, Mammoth Biosciences, BiomeSense, and Poppy Health. He is also an inventor on US patent no. 11380421, Pathogen detection using next generation sequencing, under which algorithms for taxonomic classification, filtering, and pathogen detection are used by SURPI+ software. C.Y.C. receives research support from Delve Bio and Abbott Laboratories, Inc. The other authors declare no competing interests.
References
- Popovich KJ, Snitkin ES. Whole genome sequencing—implications for infection prevention and outbreak investigations. Curr Infect Dis Rep. 2017;19:15.PubMedGoogle Scholar
- Gilchrist CA, Turner SD, Riley MF, Petri WA Jr, Hewlett EL. Whole-genome sequencing in outbreak analysis. Clin Microbiol Rev. 2015;28:541–63. DOIPubMedGoogle Scholar
- Cuomo CA. Harnessing whole genome sequencing in medical mycology. Curr Fungal Infect Rep. 2017;11:52–9.PubMedGoogle Scholar
- Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11:728–36. DOIPubMedGoogle Scholar
- Urwin R, Maiden MC. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;11:479–87.PubMedGoogle Scholar
- Benoit P, Brazer N, de Lorenzi-Tognon M, Kelly E, Servellita V, Oseguera M, et al. Seven-year performance of a clinical metagenomic next-generation sequencing test for diagnosis of central nervous system infections. Nat Med. 2024;30:3522–33.PubMedGoogle Scholar
- Bergin SP, Chemaly RF, Dadwal SS, Hill JA, Lee YJ, Haidar G, et al. Plasma microbial cell-free DNA sequencing in immunocompromised patients with pneumonia: a prospective observational study. Clin Infect Dis. 2024;78:775–84.PubMedGoogle Scholar
- Blauwkamp TA, Thair S, Rosen MJ, Blair L, Lindner MS, Vilfan ID, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol. 2019;4:663–74.PubMedGoogle Scholar
- Fourgeaud J, Regnault B, Ok V, Da Rocha N, Sitterlé É, Mekouar M, et al. Performance of clinical metagenomics in France: a prospective observational study. Lancet Microbe. 2024;5:e52–61.PubMedGoogle Scholar
- Wilson MR, Sample HA, Zorn KC, Arevalo S, Yu G, Neuhaus J, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N Engl J Med. 2019;380:2327–40.PubMedGoogle Scholar
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al.; China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–33. DOIPubMedGoogle Scholar
- Smith DJ, Gold JAW, Chiller T, Bustamante ND, Marinissen MJ, Rodriquez GG, et al.; Fungal Meningitis Response Team. Update on outbreak of fungal meningitis among U.S. residents who received epidural anesthesia at two clinics in Matamoros, Mexico. Clin Infect Dis. 2024;78:1554–8.PubMedGoogle Scholar
- US Centers for Disease Control and Prevention (CDC). Fungal meningitis outbreak associated with procedures performed under epidural anesthesia in Matamoros, Mexico [cited 2024 May 12]. https://www.cdc.gov/hai/outbreaks/meningitis-epidural-anesthesia.html
- Strong N, Meeks G, Sheth SA, McCullough L, Villalba JA, Tan C, et al. Neurovascular complications of iatrogenic Fusarium solani meningitis. N Engl J Med. 2024;390:522–9.PubMedGoogle Scholar
- Miller S, Naccache SN, Samayoa E, Messacar K, Arevalo S, Federman S, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29:831–42.PubMedGoogle Scholar
- Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–92. DOIPubMedGoogle Scholar
- Zaharia M, Bolosky B, Curtis K, Patterson D, Fox A, Patterson D, et al. Alignment in a SNAP: cancer diagnosis in the genomic age. Abstract 1912: United States and Canadian Academy of Pathology Annual Meeting; March 17–23, 2012; Vancouver, British Columbia, Canada. Lab Invest. 2012;92:458A.
- Mount DW. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007;2007:pdb.top17.
- Harrison PW, Amode MR, Austine-Orimoloye O, Azov AG, Barba M, Barnes I, et al. Ensembl 2024. Nucleic Acids Res. 2024;52(D1):D891–9.PubMedGoogle Scholar
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.PubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.PubMedGoogle Scholar
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.PubMedGoogle Scholar
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.PubMedGoogle Scholar
- Sayers EW, Cavanaugh M, Clark K, Pruitt KD, Sherry ST, Yankie L, et al. GenBank 2024 Update. Nucleic Acids Res. 2024;52(D1):D134–7.PubMedGoogle Scholar
- Galardini M, Mengoni A, Bazzicalupo M. Mapping contigs using CONTIGuator. Methods Mol Biol. 2015;1231:163–76.PubMedGoogle Scholar
- Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst Biol. 2009;58:130–45.PubMedGoogle Scholar
- Debourgogne A, Gueidan C, Hennequin C, Contet-Audonneau N, de Hoog S, Machouart M. Development of a new MLST scheme for differentiation of Fusarium solani Species Complex (FSSC) isolates. J Microbiol Methods. 2010;82:319–23.PubMedGoogle Scholar
- Chang DC, Grant GB, O’Donnell K, Wannemuehler KA, Noble-Wang J, Rao CY, et al.; Fusarium Keratitis Investigation Team. Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. JAMA. 2006;296:953–63. DOIPubMedGoogle Scholar
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017;12:1261–76. DOIPubMedGoogle Scholar
- Kilian M, Scholz CF, Lomholt HB. Multilocus sequence typing and phylogenetic analysis of Propionibacterium acnes. J Clin Microbiol. 2012;50:1158–65. DOIPubMedGoogle Scholar
- Yeo M, Mauricio IL, Messenger LA, Lewis MD, Llewellyn MS, Acosta N, et al. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl Trop Dis. 2011;5:
e1049 . DOIPubMedGoogle Scholar - O’Donnell K, Sutton DA, Fothergill A, McCarthy D, Rinaldi MG, Brandt ME, et al. Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J Clin Microbiol. 2008;46:2477–90. DOIPubMedGoogle Scholar
- Tsang AKL, Lee HH, Yiu SM, Lau SKP, Woo PCY. Failure of phylogeny inferred from multilocus sequence typing to represent bacterial phylogeny. Sci Rep. 2017;7:4536. DOIPubMedGoogle Scholar
- Bleidorn C, Gerth M. A critical re-evaluation of multilocus sequence typing (MLST) efforts in Wolbachia. FEMS Microbiol Ecol. 2018;94:1. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: April 03, 2025
Table of Contents – Volume 31, Number 5—May 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Charles Y. Chiu, UCSF China Basin Campus, 185 Berry Street, Box 0134, San Francisco, CA 94143, USA
Top