Volume 32, Number 5—May 2026
Research
Updated Genomic Epidemiologic Description of Candida (Candidozyma) auris, United States
Figure 1
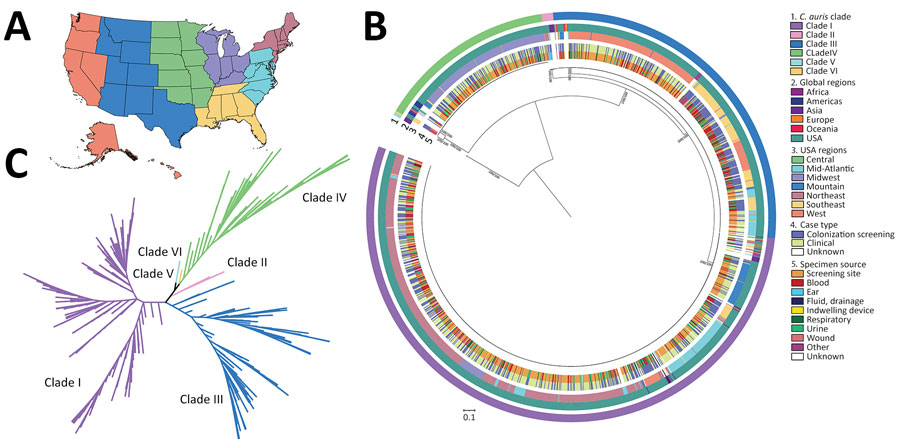
Figure 1. Phylogenetic characterization of sequenced isolates from study of genomic epidemiologic description of Candida (Candidozyma) auris in the United States. A) US map showing regions where sequenced cases originated, as defined by the Centers for Disease Control and Prevention Antimicrobial Resistance Laboratory Network (colors defined in section 3 of key at right). B) Maximum-likelihood phylogenetic tree, rooted at the midpoint, represents cases sequenced from the United States (n = 1,535) and various global regions (n = 75). C) Genetic relationships among cases represented as an unrooted phylogenetic tree colored by clade. All US cases cluster within clades I–IV. The phylogenetic trees were inferred from 421,678 whole-genome single nucleotide polymorphisms. Bootstrap support values between major clades were 100, as determined by the IQ-TREE SH-aLRT/UFboot methods.