Volume 6, Number 2—April 2000
Synopsis
Vaccines for Mucosal Immunity to Combat Emerging Infectious Diseases
The Mucosal Immune System
T cell and Cytokine Involvement in B-Cell Isotype Commitment to IgA
Mucosal S-IgA Antibody Responses
Cell-Mediated Immunity at Mucosal Surfaces
Antigen-Specific CTLs
CTLs in the Intestinal Tract
CTLs in the Respiratory and Urogenital Tracts
The Common Mucosal Immune System
Mucosal Vaccines
Future Directions
Cite This Article
Cite This Article
Citation for Media
Abstract
The mucosal immune system consists of molecules, cells, and organized lymphoid structures intended to provide immunity to pathogens that impinge upon mucosal surfaces. Mucosal infection by intracellular pathogens results in the induction of cell-mediated immunity, as manifested by CD4-positive (CD4+) T helper-type 1 cells, as well as CD8+ cytotoxic T-lymphocytes. These responses are normally accompanied by the synthesis of secretory immunoglobulin A (S-IgA) antibodies, which provide an important first line of defense against invasion of deeper tissues by these pathogens. New-generation live, attenuated viral vaccines, such as the cold-adapted, recombinant nasal influenza and oral rotavirus vaccines, optimize this form of mucosal immune protection. Despite these advances, new and reemerging infectious diseases are tipping the balance in favor of the parasite; continued mucosal vaccine development will be needed to effectively combat these new threats.
Behavioral changes in the human host may be contributing to the emergence of new diseases. Perhaps emerging pathogens become resistant to antibiotics or (through genetic recombination) become more resistant to host defenses. Recombination events or lack of exposure can result in loss of immunity of the population to the pathogen, as has been well documented with influenza virus. Recombination events increase the infection rate by the emerging pathogen and, in the case of influenza virus, occasionally result in pandemics. A large number of emerging pathogens are mucosally transmitted and must cross mucosal barriers to infect the host. Thus, our ultimate defense against new and reemerging infectious disease will require effective mucosal vaccines.
Mucosal surfaces are prominent in the gastrointestinal, urogenital, and respiratory tracts and provide portals of entry for pathogens. Increased sanitation and hygiene, the use of antibiotics, and childhood vaccination have enormously decreased the death rate from infectious diseases over the last century (1). Thus, infectious agents not controlled by antibiotics and improved sanitation and hygiene measures would most likely be prevalent under current conditions. Pathogens in this category include respiratory viruses, for example, influenza virus. Another group includes sexually transmitted disease (STD) pathogens, for example, HIV. The fact that a) pneumonia and influenza virus and b) HIV infection were listed as the leading causes of death (ranked number 6 and 8, respectively) by infectious agents in the United States in 1997 confirms this notion (1). However, other viral and bacterial STDs are of major concern.
The best defense against these predominantly mucosal pathogens would be vaccines, preferably mucosal vaccines capable of inducing both systemic and mucosal immunity. Although numerous strategies exist for the induction of mucosal immune responses, we will focus on the use of live attenuated vectors, such as Salmonella typhi and adenovirus. In a recent National Institutes of Health news release (April 8, 1999) from the Institute of Medicine report on domestic vaccine priorities for the future, which compared costs and health benefits, influenza was listed as one of seven diseases requiring an effective vaccine. The induction of protective mucosal immunity to influenza virus has made considerable progress with the development of cold-adapted influenza strains, which are in phase-3 clinical trials. Mucosal immunity, which is important for long-term protection, forms a first line of defense against mucosally transmitted pathogens such as influenza. Mucosal defense against pathogens consists of both innate barriers, such as mucous, epithelium, and innate immune mechanisms, and adaptive host immunity, which at mucosal surfaces consists predominantly of CD4+ T cells, secretory immunoglobulin A (S-IgA), and antigen-specific cytotoxic T-lymphocytes (CTLs). This review will focus on the antigen-specific mucosal immune system.
The major obstacle in combating emerging infectious diseases is the lack of more effective antibiotics and vaccines. Misuse of antibiotics has led to antibiotic-resistant pathogens, further intensifying the need for mucosal vaccine development--a cost-effective disease-prevention tool. The Department of Vaccines and Other Biologicals within the World Health Organization (WHO) has defined several priority vaccines for accelerated introduction; they include pneumococcal, Haemophilus influenzae type b, rotavirus, and hepatitis B. Other vaccine goals within WHO are eradication of polio, reduction of measles cases by 90%, and elimination of neonatal tetanus (2). For most pathogens targeted by WHO for vaccines, induction of mucosal immunity appears most appropriate based on the routes of infection. Thus, a better understanding of the mucosal immune system is needed before effective mucosal vaccines can be developed.
Figure 1
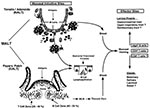
Figure 1. M cells and the induction of mucosal immunity. M cells are present in mucosal inductive sites in both the intestinal and upper respiratory tract, specifically in Peyers patches and the nasal-associated...
Mucosal inductive sites in humans, such as the Peyers patches in the intestinal tract and the nasal-associated lymphoreticular tissue in the oropharyngeal cavity, stand as sentinels to the intestinal and respiratory tracts and represent the major sites where mucosal immune responses are initiated. Common features of these inductive sites are microfold or M cells. Although the precise function of M cells has not yet been established, recent studies indicate that they are involved in uptake, transport, processing, and possibly presentation of microbial antigens (3,4). The interaction of epithelial cells with T and B lymphocytes induces epithelial cells to differentiate into M cells in vitro (5), indicating the importance of lymphocyte-epithelial cell interactions for maintaining M cells in the follicle-associated epithelium of the Peyers patches (Figure 1). These lymphocyte-M cell interactions can occur in the pocket of the M cells and are mediated through thin cellular extensions, indicating that cell-cell interactions are an intricate part of the M-cell function and that they may facilitate transfer of luminal antigens, viruses, bacteria, and other protein components (3). The ability of the M cell to transport particulates from the lumen across the epithelial barrier has been exploited by some pathogens to facilitate entry into the host, as has been demonstrated for invasive strains of Salmonella (6) and reovirus (7) (Figure 1). Identification of bacterial and viral virulence factors associated with targeting M cells, such as the sigma protein from reovirus, may allow development of mucosal vaccines and vectors to deliver vaccine components directly into mucosal inductive sites.
Although the variables involved in the switching of B cells to polymeric IgA (pIgA)-producing plasma cells have been studied, many questions remain. In recent years, gene-deleted or knockout mice have contributed to a better understanding of the role of specific cells, cytokines, and surface molecules involved in IgA isotype switching. Presumably, isotype switching occurs in mucosal inductive sites, while IgA production by plasma cells occurs in mucosal effector sites, separating the IgA switching and IgA secretion by B cells into different immune compartments (8). Each of these stages requires specific signals, such as costimulatory molecules, cytokines, and T-helper cells, to give rise to antigen-specific S-IgA Abs in mucosal effector sites.
Figure 2
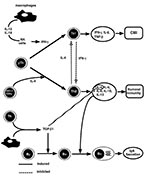
Figure 2. Differentiation and regulation of T-helper subsets and the immune response in the mucosal compartments. Encounter of pathogen-derived antigen or vaccine antigen will stimulate T-helper cells to secrete cytokines. Depending on the...
Neither Th1- nor Th2-type cytokines contributed significantly to the switching of B cells to surface IgA positive (sIgA+) B cells. This process required the presence of transforming growth factor bð1 (TGF-bð1) (Figure 2), which can activate the switch of B cells to the IgA isotype (9). TGF-bð1 induces a small proportion (<2%) of B cells to switch to IgA in activated B-cell cultures (9,10). However, TGF-bð1, when used in combination with additional signals, increased TGF-bð1-induced switching in 10% to 20% of B cells and approached IgA+ B-cell levels observed in Peyers patches (11). Thus, multiple activation signals contribute to the switch to IgA, i.e., B-cell activation by cross-linking the B-cell antigen receptor, CD40-CD40L interactions to promote switching, TGF-bð1 by directing the switch to IgA, and Th2-type cytokines by increasing the number of post-switch IgA+ B cells and their differentiation into IgA-secreting plasma cells. In addition, activated T cells and dendritic cells from the Peyers patches were more effective in switching sIgM+sIgA- B cells to IgA-producing cells than were T cells and dendritic cells derived from the spleen (12). This suggests that mucosal inductive sites contain specialized T cells or dendritic cells beneficial for B cells to differentiate into IgA-producing cells.
T-cell helper functions play important roles in generating antigen-specific humoral and cell-mediated immunity in both systemic and mucosal compartments. The importance of CD4+ T cells for generating protective immunity is illustrated by the lack of these cells in AIDS patients. The differentiation of Th0 cells into either Th1 or Th2 is driven by cytokines such as interleukin 12 (IL-12), interferon gð (IFN-gð), and IL-4, respectively. For example, intracellular pathogens, such as viruses and intracellular bacteria, induce production of IL-12 or IL-18 by activated macrophages, presumably after ingestion of the partuculate pathogen, inducing IFN-gð production in natural killer (NK) cells, which in turn drives the differentiation of Th0 cells toward a Th1 phenotype producing IFN-gð, IL-2, and tumor necrosis factor bð (TNF-bð)ð (Figure 2). Murine Th1-type responses are associated with cell-mediated immunity, such as delayed-type hypersensitivity and IgG2a antibody responses (8). Th0 cells are differentiated into Th2-type cells when soluble exogenous antigen is administered, triggering CD4+, NK1.1+ T cells to produce IL-4. The Th2 cell produces more IL-4, expanding Th2-cells, which support the associated immune response. Th2 cells secrete cytokines such as IL-4, IL-5, IL-6, IL-9, IL-10, IL-13. The production of IL-4 supports IgG1 subclass and IgE production, but other antibodies such as IgG2b and IgA, are also produced during a Th2-dominated response (8).
It is not known whether Th1 or Th2 cells are beneficial for optimal S-IgA production. Historically, Th2-type cytokines were considered major helpers for antibody responses. For example, S-IgA Ab responses were supported by mucosal adjuvants such as cholera toxin, which induced polarized Th2 cell responses (13). However, S-IgA Ab responses may also be induced through Th1-dominated responses, as observed with intracellular pathogens such as Salmonella in the gastrointestinal tract (14) or influenza virus in the upper respiratory tract (15). Thus, either Th1 or Th2 cells or a combination of these cell types can support antigen-specific S-IgA Ab responses. In this respect, Th2-type cytokines play a role in terminal differentiation of B cells, that are already committed to IgA (16-18), while the Th1-type cytokine IFN-gð has been implicated in the induction of the polymeric Ig receptor (pIgR) needed for transport of S-IgA (19). Cross-inhibition of Th1 and Th2 cell-directed IgG2a and IgE production was mediated through IFN-gð and IL-4, respectively (Figure 2) (20,21).
The hallmark of the mucosal immune system is the production of S-IgA. S-IgA results from transcytosis of pIgA across the epithelium through binding to the pIgR. S-IgA is released from the pIgR by cleavage of the receptor, resulting in pIgA covalently associated with a substantial part of the pIgR, i.e., the secretory component (22). This complex, referred to as S-IgA, seems to be more resistant to proteolysis in external secretions. Additional roles for S-IgA in protection are suggested by its reduction of influenza virus attachment and its prevention of internalization of virus into baby hamster kidney cells. In contrast, the action of monomeric IgA is indistinguishable from that reported for IgG and is less efficient than S-IgA for inhibition of influenza virus entry (23). In addition, pIgA, as opposed to IgG or monomeric IgA, neutralizes virus intracellularly, as first was shown with Sendai virus (24). Furthermore, transport of pIgA containing immune complexes across epithelial cells expressing the pIgR is another defense mechanism of the mucosal immune system against pathogen entry (25).
These characteristics of pIgA are beneficial in preventing infection and inflammation at epithelial surfaces. The transport of pIgA across epithelial cells allows active elimination of immune complexes at mucosal sites and even virus inside epithelial cells. Evidence that these observations were not an artifact of the in vitro system was provided by the murine backpack model of rotavirus-specific monoclonal antibodies (mAb) to VP4 and VP6 proteins. In this model, nonneutralizing VP6-specific IgA mAb were protective, but not when administered directly to the gastrointestinal tract, indicating that IgA transcytosis played a prominent role in effective immune exclusion (26). Thus, virus-specific, intra-epithelial IgA can inhibit viral entry and replication.
Although S-IgA has been shown to be an important effector molecule to protect mucosal surfaces, the contribution to mucosal protection by the cellular immune system should not be underestimated. The strategic advantage of cell-mediated versus antibody-mediated immune responses is that T cells can recognize peptides derived from core proteins of the pathogen, such as influenza virus. Core proteins are usually expressed and presented much earlier during infection than proteins targeted for neutralizing antibodies, such as HA and NA of influenza virus. Subsequently, cell-mediated immunity (CMI) occurs before the induction of antibodies and forms an early line of defense. Although antibodies to core proteins are also formed later in the immune response, their exact role in protective immunity is not clear. Besides supporting humoral immunity, CD4+ T-helper cells function in CMI as producers of cytokines, which mediate delayed-type hypersensitivity and support CTLs and which as such are critical components of the CMI responses to intracellular pathogens. For example, major histocompatibility complex (MHC)-restricted CTL responses are supported by Th1 cells. Cytotoxic cells can be classified based on antigen specificity and MHC restriction, i.e., nonspecific cytotoxic cells and antigen-specific, MHC-restricted CTLs. The first kind is composed of various cell types, including NK cells and antibody-dependent cytotoxicity, and functions very early in the immune response (day 1 to 3), and these cells are detected through out the mucosal immune system. Presumably, they decrease pathogen load in the early stage of the immune responses, while antigen-specific responses are still being established. The second type, antigen-specific CTL, achieved optimal activity a little later than nonspecific CTL, i.e., at day 3 to 5 of the immune response before antibody production.
Figure 3
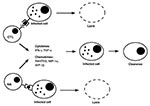
Figure 3. Pathways of intracellular pathogen clearance from infected cells by cytotoxic cells. Intracellular pathogen-derived antigens complexed to MHC class I molecules are recognized by CTLs, while NK cells recognize the absence or...
Both antigen-specific and nonspecific cytotoxic cell types can control growth of intracellular pathogens by two distinct mechanisms (Figure 3). First, they can respond to the infection by secreting a number of cytokines such as IFN-gð and TNF-að (27) or chemokines such as Rantes, macrophage-inflammatory protein-1að (MIP-1að)ð, and MIP-1bð (28,29). These soluble factors inhibit growth of intracellular pathogens such as viruses without destroying the host cell. Second, cytotoxic cells can effectively and efficiently recognize and lyse infected cells and prevent multiplication of viruses. Little is known about the induction, compartmentalization, and homing pattern of cytotoxic cells. Their presence in the mucosal compartment upon infection reflects their importance for protection against pathogens at mucosal surfaces.
CTLs play an important role in the elimination of cells infected with various intracellular pathogens by recognizing pathogen-specific antigen/MHC complexes. Antigen-specific CTLs inhibit further spread of pathogens and help to terminate infections. Compartmentalization of pathogen-specific CTL responses has been reported and located at the site of initial infection. For example, CTLs preferentially compartmentalize in mucosa-associated lymphoreticular tissues after pulmonary or intestinal infection.
Presentation of rotavirus by the intestinal mucosal surface was not required for induction of virus-specific cytotoxic inter-epithelial lymphocytes in the intestinal tract (30). In addition, the site at which rotavirus is first presented to the immune system will determine the site where rotavirus-specific CTL precursors (CTLp) first appear; however, regardless of the route of inoculation, rotavirus-specific CTLp can be found throughout the lymphoid system 21 days after the initial infection (31). Adoptive transfer of splenic lymphocytes from immunized animals protected suckling mice against murine rotavirus-induced gastroenteritis in the absence of rotavirus-specific neutralizing antibodies, indicating that antigen-specific CTLs protect against mucosal pathogens in the intestinal tract (32). Thus, thymus-derived aðbð T cells can migrate to the intestinal epithelium after antigen-specific activation and protect the host against subsequent challenge. This notion is supported by findings that systemic immunization with attenuated macaque-specific Simian immunodeficiency virus induced virus-specific CTL responses in gut-associated lymph nodes and limited superinfection following mucosal challenge (33).
The distribution of CTLs following influenza virus infection in different mucosal compartments indicates that lymph nodes draining mucosal surfaces function as reservoirs for memory T cells. Mediastinal lymph nodes, the draining lymph nodes of the lungs, are considered the site where antigen presentation to T cells initially occurs before clonal expansion. Subsequently, T cells migrate to effector sites (mesenchyma of the lungs and airways) to interact with infected cells (34,35). Thus, compartmentalization of memory CTL responses to mucosa-associated lymphoid tissue may be related to the initial site of virus infection. This notion was confirmed by the observation that induction of protective antiviral memory CTL in mucosal tissues depends on region-specific mucosal immunization (36). HIV-specific CTL have been detected in the cervix (37) or semen (38) of HIV-infected persons. These studies indicate that the initial site of antigen exposure and induction of antigen-specific CTL responses in the urogenital tract are associated. Further evidence for this notion comes from the use of MHC class 1 tetramere technology, by which antigen-specific quantitation of CD8+ T cells can be performed. Upon intranasal influenza administration, most antigen-specific, IFN-gð-producing effector CD8+ T cells were located in bronchial lavages and that both effector (eCTL) and memory CTL (mCTL) occurred at a much higher frequency than initially thought based on limiting dilution assays (39). Since respiratory virus infection induces enlargement of the mediastinal lymph nodes early in the immune response and since these lymph nodes contain a relative small number of mCTL after initial exposure, a strong recruitment from circulating T cells occurs, or alternatively, clonal expansion of the resident mCTL takes place (39).
The presence of CTLs in mucosal compartments may contribute to the control of, and recovery from, infection by intracellular pathogens at mucosal surfaces. Since different pathogens have distinct infection routes or different localization in the host, compartmentalization of protective, antigen-specific CTLs may vary, based on the specific pathogen. In general, mucosal infection induces primarily antigen-specific CTLs in the mucosal compartment and mucosa-associated lymphoid organs and depends on mucosal infection to control pathogens at the port of entry, i.e., the mucosal surfaces.
Antigenic exposure at mucosal sites activates mucosal B and T-lymphocytes to emigrate from the inductive site and home to various mucosal effector sites. The common mucosal immune system involves homing of antigen-specific lymphocytes to mucosal effector sites other than the site where initial antigen exposure occurred. This pathway has almost exclusively been documented for S-IgA antibody responses at mucosal surfaces mediated by B cells, but similar events are assumed to take place with T cells. Different immunization routes, such as oral, rectal, and intranasal, can induce generalized mucosal immune responses. However, oral immunization induced a more restricted mucosal response, as reflected by a more restricted homing receptor profile than nasal immunization. Specifically, after systemic immunization the predominant homing receptor on antibody secreting cells is the L selectin, after oral immunization the að4bð7 integrin, and after nasal immunization a large portion expressed both the L selectin and the að4bð7 integrin. The fact that nasal immunization induced antibodies in a broader range of tissues, such as saliva and the urogenital tract, than oral immunization reflects the more restricted nature of oral immunization (8).
Circumstantial evidence indicates the existence of a common mucosal immune system for cell-mediated immunity (44). The data available indicate that antigen-specific CTL responses at mucosal surfaces are dictated by induction of CTL locally and are not due to migration from distant sites. CTL do normally migrate to the systemic compartment. It could be hypothesized that the presence of antigen-specific CTL in the systemic compartment would allow for quick, protective responses at any mucosal site, but more research is needed to confirm this hypothesis. Such research is crucial, since limited CTL activity at mucosal surfaces could be a built-in mechanism to protect the mucosal epithelium from damage, a notion supported by the observation that pCTL in immunologically privileged sites fail to differentiate into fully functional CTL, unless exposed to antigen (40). This concept could have a major influence on future vaccine development. If mucosal antigen-specific memmory CTL responses are observed only after mucosal immunization, optimal protection against pathogens would require the use of mucosal vaccine. However, systemic induced CTL can generate an antigen-specific mucosal CTL response; in addition, systemic immunization can be used for cell-mediated protection at mucosal surfaces.
Although mucosal application of vaccines is attractive for many reasons, only a few mucosal vaccines, mostly oral, have been approved for human use. These vaccines include poliovirus, Salmonella typhi, and the recently approved tetravalent rotavirus vaccine, RotaShield, consisting of reassorted rhesus-human rotaviruses. The latter vaccine has recently been associated with intussusception, a type of bowel obstruction, and its use has temporarily been suspended to await a more detailed analysis of this potential problem. Human approved mucosal vaccines so far involve live attenuated pathogens, and for this reason oral poliovirus vaccine is recommended after receiving the injected inactivated virus, since a limited number of polio cases occur after immunization with the live attenuated virus. Since the live virus induces better, longer-lasting protection, it is given after some level of systemic immunity has been achieved, to limit possible problems. Another oral vaccine is the typhoid fever vaccine, which consists of attenuated S. typhi strain Ty21a. The cold-adapted influenza virus (CAIV), which is in advanced clinical trials, is the first mucosal vaccine given nasally to humans and has been shown to generate protective immune responses (41). Thus, CAIV are promising vehicles for generating protective immunity to influenza in children. The use of CAIV may also resolve some of the problems observed during the recent outbreak of the Hong Kong virus. Due to its relatedness with A/Chicken/Hong Kong/258/97 (H5N1) virus, the production of this virus for vaccine purposes was severely hampered because of its lethal effect on chicken eggs. The use of CAIV, which is readily produced by reassortment, might overcome this problem and allow production of high-titer virus for vaccine purposes.
An alternative approach is the use of DNA vaccines. Plasmid DNA was used in clinical trials to induce protection against several pathogens, including hepatitis B virus, herpes simplex virus, HIV, malaria, and influenza (42). However, in all cases induction of antibodies and CTL in the systemic but not the mucosal compartment were reported. Although some progress has been made in inducing mucosal immunity in laboratory animals with DNA vaccines by using cationic lipids or other delivery vehicles, as well as immunostimulatory CpG dinucleotide motifs, no reports exist on the induction of mucosal immunity by DNA vaccines in humans. Unmethylated CpG dinucleotides are immunostimulatory, especially when presented in a 6 base-pair motif in which the central CpG is flanked by two 5' purines and two 3' pyrimidines.
Another promising avenue for mucosal vaccines is the bacterial adhesins. Mucosal antibodies to these proteins block the pathogen's ability to penetrate mucosal barriers. Adhesins are very attractive options because of the highly conserved nature of these proteins due to their association with conserved host receptor proteins. The pilus-associated adhesin FimH from uropathogenic E. coli binding to mannose-oligosaccharides is a vaccine target. Mucosally administered vaccines containing FimH are in clinical trials that will assess their efficacy compared with parenterally administered vaccines. Furthermore, the recently approved acellular pertussis vaccine also contains adhesins, i.e., the filamentous hemagglutinin and pertactin, which recognize sulphated sugars on glycoconjugates and the integrin-binding protein motif Arg-Gly-Asp, respectively. This indicates that adhesin-specific immunity might be a successful approach for generating mucosal protection against pathogens (43). The importance of blocking the initial attachment and entry into the host cell has been recognized for some time for viruses such as influenza, but the use of this approach for bacteria, still in its infancy, has enormous potential for mucosal vaccines.
The mucosal immune system is a complex and redundant system that generates large amounts S-IgA as well as cell-mediated immunity at mucosal surfaces to prevent pathogen infiltration and inflammation. The mucosal immune system should be most efficient in providing protection against pathogens and generating longer-lasting protection through using attenuated pathogens for vaccines purposes. The only mucosal vaccines approved for humans are attenuated pathogens. Future mucosal vaccines will also involve vaccine strategies other than attenuated pathogens. For example, DNA vaccines or subunit vaccines, such as bacterial adhesins, in combination with potent mucosal adjuvants (such as QS21 a saponin derived from the bark of the South American tree Quillaja saponia Molina, mutant enterotoxins, unmethylated CpG motifs, or cytokines such as IL-12) or mucosal delivery systems, such as microspheres, will have the potential to be the next generation of vaccines inducing mucosal protection to pathogens in humans.
Dr. van Ginkel is assistant professor of microbiology at the University of Alabama at Birmingham. His research interests focus on the mucosal immune response in the respiratory tract, with an emphasis on viral immunity.
Acknowledgments
We thank Kimberly McGhee for editorial assistance on this manuscript.
The work was supported by National Institutes of Health grants P30 DK 54781, AI 18958, AI 43197, DK 44240, T32 AI 07150 and contracts N01 65298 and N01 65299.
References
- V&B annual report, 1998. Department of vaccines and other biologicals. Geneva: World Health Organization; 1999.
- Neutra MR, Frey A, Kraehenbuhl J-P. Epithelial M cells: gateways for mucosal infection and immunization. Cell. 1996;86:345–8. DOIPubMedGoogle Scholar
- Allan CH, Mendrick DL, Trier JS. Rat intestinal M cells contain acidic endosomal-lysosomal compartments and express class II major histocompatibility complex determinants. Gastroenterology. 1993;104:698–708.PubMedGoogle Scholar
- Kerneis S, Bogdanova A, Kraehenbuhl J-P, Pringault E. Conversion by Peyer's patch lymphocytes of human enterocytes into M cells that transport bacteria. Science. 1998;277:949–52. DOIGoogle Scholar
- Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J Exp Med. 1994;180:7–9. DOIPubMedGoogle Scholar
- Wolf JL, Rubin DH, Finberg R, Kauffman RS, Sharpe AH, Trier JS, BN Intestinal M cells: a pathway for entry of reovirus into the host. Science. 1981;212:471–2. DOIPubMedGoogle Scholar
- McGhee JR, Lamm ME, Strober W. Mucosal immune responses: an overview. In: Mucosal immunology. Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, eds. San Diego: Academic Press; 1999. p. 485-506.
- Ehrhardt RO, Strober W, Harriman GR. Effect of transforming growth factor (TGF)-bð1 on IgA isotype expression. TGF-bð1 induces a small increase in sIgA+ B cells regardless of the method of B cell activation. J Immunol. 1992;148:3830–6.PubMedGoogle Scholar
- Coffman RL, Lebman DA, Shrader B. Transforming growth factor-bð specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039–44. DOIPubMedGoogle Scholar
- McIntyre TM, Kehry MR, Snapper CM. Novel in vitro model for high-rate IgA class switching. J Immunol. 1995;154:3156–61.PubMedGoogle Scholar
- Spalding DM, Williamson SI, Koopman WJ, McGhee JR. Preferential induction of polyclonal IgA secretion by murine Peyer's patch dendritic cell T cell mixtures. J Exp Med. 1984;160:941–6. DOIPubMedGoogle Scholar
- Xu-Amano J, Kiyono H, Jackson RJ, Staats HF, Fujihashi K, Burrows PD, Helper T cell subsets for immunoglobulin A responses: oral immunization with tetanus toxoid and cholera toxin as adjuvant selectively induces Th2 cells in mucosal associated tissues. J Exp Med. 1993;178:1309–20. DOIPubMedGoogle Scholar
- VanCott JL, Staat HF, Pascual DW, Roberts M, Chatfield SN, Yamamoto M, Regulation of mucosal and systemic antibody responses by Thelper subsets, macrophages, and derived cytokines following oral immunization with live recombinant Salmonella. J Immunol. 1996;156:1504–14.PubMedGoogle Scholar
- Novak M, Yamamoto M, Fujihashi K, Moldoveanu Z, Kiyono H, McGhee JR, Ig-secreting and interferon-gamma-producing cells in mice mucosally immunized with influenza virus. Adv Exp Med Biol. 1995;371B:1587–90.PubMedGoogle Scholar
- Briere F, Bridon JM, Chevet D, Souillet G, Bienvenu F, Guvet C, Interleukin 10 induces B lymphocytes from IgA-deficient patients to secrete IgA. J Clin Invest. 1994;94:97–104. DOIPubMedGoogle Scholar
- Fujihashi K, McGhee JR, Lue C, Beagley KW, Taga T, Hirano T, Human appendix B cells naturally express receptors for and respond to interleukin 6 with selective IgA1 and IgA2 synthesis. J Clin Invest. 1991;88:248–52. DOIPubMedGoogle Scholar
- Lebman DA, Nomura DY, Coffman RL, Lee FD. Molecular characterization of germline immunoglobulin A transcripts produced during transforming growth factor bð-induced isotype switching. Proc Natl Acad Sci U S A. 1990;87:3962–6. DOIPubMedGoogle Scholar
- Wira CR, Richardson J, Prabhala R. Endocrine regulation of mucosal immunity: effect of sex hormones and cytokines on the afferent and efferent arms of the immune system in the female reproductive tract. In: Ogra PL, Mestecky J, Lamm ME, McGhee JR, Strober W, Bienenstock J, editors. Handbook of mucosal immunology. San Diego: Academic Press; 1994. p. 705-16.
- Rizzo LV, DeKruyff RH, Umetsu DT, Caspi RR. Regulation of the interaction between Th1 and Th2 T cell clones to provide help for antibody production in vivo. Eur J Immunol. 1995;25:708–16. DOIPubMedGoogle Scholar
- DeKruyff RH, Rizzo LV, Umetsu DT. Induction of immunoglobulin synthesis by CD4+ T cell clones. Semin Immunol. 1993;5:421–30. DOIPubMedGoogle Scholar
- Mostov KE. Transepithelial transport of immunoglobulins. Annu Rev Immunol. 1994;12:63–84. DOIPubMedGoogle Scholar
- Taylor HP, Dimmock NJ. Mechanism of neutralization of influenza virus by secretory IgA is different from that of monomeric IgA or IgG. J Exp Med. 1985;161:198–209. DOIPubMedGoogle Scholar
- Mazanec MB, Kaetzel CS, Lamm ME, Fletcher D, Nedrud JG. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc Natl Acad Sci U S A. 1992;89:6901–5. DOIPubMedGoogle Scholar
- Kaetzel CS, Robinson JK, Lamm ME. Epithelial transcytosis of monomeric IgA and IgG cross-linking through antigen to polymeric IgA. A role for monomeric antibodies in the mucosal immune response. J Immunol. 1994;152:72–6.PubMedGoogle Scholar
- Burns JW, Siadat-Pajouh M, Krishnaney AA, Greenberg HB. Protective effects of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science. 1996;272:46–8. DOIPubMedGoogle Scholar
- Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Invest. 1997;99:1472–7. DOIPubMedGoogle Scholar
- Fehniger TA, Herbein G, Yu H, Para MI, Bernstein ZP, O'Brien WA, Natural killer cells from HIV-1+ patients produce C-C chemokines and inhibit HIV-1 infection. J Immunol. 1998;161:6433–8.PubMedGoogle Scholar
- Yang OO, Kalams SA, Trocha A, Cao H, Luster A, Johnson RP, Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J Virol. 1997;71:3120–8.PubMedGoogle Scholar
- Offit PA, Dudzik KI. Rotavirus-specific cytotoxic T lymphocytes appear at the intestinal mucosal surface after rotavirus infection. J Virol. 1989;63:3507–12.PubMedGoogle Scholar
- Offit PA, Cunningham SL, Dudzik KI. Memory and distribution of virus-specific cytotoxic T lymphocytes (CTLs) and CTL precursors after rotavirus infection. J Virol. 1991;65:1318–24.PubMedGoogle Scholar
- Offit PA, Dudzik KI. Rotavirus-specific cytotoxic T lymphocytes passively protect against gastroenteritis in suckling mice. J Virol. 1990;64:6325–8.PubMedGoogle Scholar
- Cranage MP, Whatmore AM, Sharpe SA, Cook N, Polyanskaya N, Leech S, Macaques infected with live attenuated SIVmac are protected against superinfection via the rectal mucosa. Virology. 1997;229:143–54. DOIPubMedGoogle Scholar
- Hou S, Doherty PC. Partitioning of responder CD8+ T cells in lymph node and lung of mice with Sendai virus pneumonia by LECAM-1 and CD45RB phenotype. J Immunol. 1993;150:5494–500.PubMedGoogle Scholar
- Carding SR, Allan W, McMickle A, Doherty PC. Activation of cytokine genes in T cells during primary and secondary murine influenza pneumonia. J Exp Med. 1993;177:475–582. DOIPubMedGoogle Scholar
- Gallichan WS, Rosenthal KL. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med. 1996;184:1879–90. DOIPubMedGoogle Scholar
- Musey L, Hu Y, Eckert L, Christensen M, Karchmer T, McElrath MJ. HIV-1 induces cytotoxic T lymphocytes in the cervix of infected women. J Exp Med. 1997;185:293–303. DOIPubMedGoogle Scholar
- Quayle AJ, Coston WMP, Trocha AK, Kalams SA, Mayer KH, Anderson DJ. Detection of HIV-1-specific CTLs in the semen of HIV-infected individuals. J Immunol. 1998;161:4406–10.PubMedGoogle Scholar
- Flyn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–91. DOIPubMedGoogle Scholar
- Ksander BR, Streilein JW. Failure of infiltrating precursor cytotoxic T cells to acquire direct cytotoxic function in immunologically privileged sites. J Immunol. 1990;145:2057–63.PubMedGoogle Scholar
- Gruber WC, Belshe RB, King JC, Treanor JJ, Piedra PA, Wright PA, Evaluation of live attenuated influenza vaccines in children 6-18 months of age: safety, immunogenicity, and efficacy. J Infect Dis. 1996;173:1313–9.PubMedGoogle Scholar
- Wizemann TM, Adamou JE, Langermann S. Adhesins as targets for vaccine development. Emerg Infect Dis. 1999;5:395–403. DOIPubMedGoogle Scholar
- Gallichan WS, Rosenthal KL. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med. 1996;184:1879–90. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 6, Number 2—April 2000
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Frederik W. van Ginkel, Department of Microbiology, University of Alabama at Birmingham, BBRB Room 775, 845 19th Street South, Birmingham, AL 35294, USA; fax: 205-975-4431
Top