Volume 7, Number 3—June 2001
Synopsis
Spoligotype Database of Mycobacterium tuberculosis: Biogeographic Distribution of Shared Types and Epidemiologic and Phylogenetic Perspectives
Cite This Article
Citation for Media
Abstract
We give an update on the worldwide spoligotype database, which now contains 3,319 spoligotype patterns of Mycobacterium tuberculosis in 47 countries, with 259 shared types, i.e., identical spoligotypes shared by two or more patient isolates. The 259 shared types contained a total of 2,779 (84%) of all the isolates. Seven major genetic groups represented 37% of all clustered isolates. Two types (119 and 137) were found almost exclusively in the USA and accounted for 9% of clustered isolates. The remaining 1,517 isolates were scattered into 252 different spoligotypes. This database constitutes a tool for pattern comparison of M. tuberculosis clinical isolates for global epidemiologic studies and phylogenetic purposes.
In 1997, 8 million new cases of tuberculosis (TB) were reported worldwide; 3.5 million cases were considered highly contagious (1). With Africa and some countries having up to 20% of their populations infected with HIV, AIDS will have a major impact on TB in coming years (2). Emergence of multidrug- resistant (MDR) strains of Mycobacterium tuberculosis is also of great epidemiologic concern (3). In this context, molecular fingerprinting of M. tuberculosis complex isolates is a powerful tool that permits detection of transcontinental spread of TB (4) and outbreaks (5). Our laboratory has described a preliminary spoligotyping database that suggested the biogeographic specificity of some of the spoligotypes from the Caribbean (6). The initial aim of this work was twofold. First, such an inventory was mandatory to detect and estimate the relative importance of TB of foreign origin in the French Caribbean. Although the incidence of TB in Martinique and Guadeloupe is comparable with that in metropolitan France (approximately 10/100,000 new cases each year), this region is part of an area of Latin America and the Caribbean with high TB prevalence. Second, we used spoligotyping results to infer potential phylogenetic relationships of M. tuberculosis strains in the Caribbean region and the history of TB by using molecular markers. An updated database could also be helpful in developing new statistical approaches in the field of population genetics of circulating M. tuberculosis clinical isolates.
Figure A1

By systematically analyzing published spoligotypes, we have now collected 3,319 spoligotyping patterns of various origins in a single database, essentially from Europe and the USA (Table 1). This database includes 259 shared types containing 2 to 476 patterns (Figure A1). The main database also includes 540 "orphan patterns" (clinical isolates showing a unique spoligotype), for a current total of 799 distinct spoligotype patterns. This article describes the nomenclature and phylogenetic reconstruction of these 259 shared types.
Spoligotyping based on the variability of the Direct Repeat (DR) locus and analysis of a variable number of tandem DNA repeats (VNTR) of M. tuberculosis were performed according to the original protocols (7,8). For the construction of the database, spoligotyping results were entered into Excel spreadsheet files in chronological order, according to the availability of results from published articles and our own investigations. The database was searched regularly for new shared types, i.e., identical spoligotypes shared by two or more patient isolates. For phylogenetic reconstruction, the spoligotyping results were entered into Recognizer software of the Taxotron package (Taxolab, Institut Pasteur, Paris), as recommended (9). The "1-Jaccard" Index was calculated for each pairwise comparison of patterns (10), and the neighbor-joining algorithm was used for building trees (11).
The source of the data and its representativeness are shown in Table 1. Of 3,319 individual spoligotypes in our database, most (2,418 [73%]) were either from Europe (1,142 [34%]) or the USA (1,283 [39%]). Spoligotypes shared between the USA and Europe totaled 1,286 isolates distributed among 45 shared types (Europe, n=461; USA, n=825). A statistical analysis was performed for the 1,286 isolates to evaluate the biogeographic specificity of the shared types and assess potential sampling bias by using a sample homogeneity test derived from the chi-square test (see below).
Description of Database
The 3,319 spoligotypes were grouped into 259 shared types containing 2,779 (84%) of the isolates and 540 (16%) orphan spoligotyping patterns (clinical isolates showing a unique spoligotype; results not shown; see Figure A1). This gives a current total of 799 distinct spoligotype patterns in our database.
Figure 1
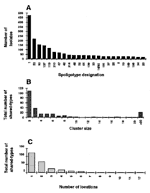
Figure 1. . Histograms derived from database (online) summarizing the distribution of shared types (A), their respective sizes (B), and their relative distribution in different locations (C).
The distribution of shared types, their respective sizes, and their relative distribution in different locations (distinct countries or geographic regions) are summarized in Figure 1. The 24 most frequent shared types totaled 1,804 (65%) isolates (Figure 1A); 7 types were highly frequent, representing 1,250 (45%) isolates. The Beijing type (type 1) was most frequent and represented 18% of isolates. Two types (119 and 137), which were almost exclusively found in the USA, accounted for 9% of isolates and may be specific for American populations or outbreaks (12). Types 53 and 50 accounted for 8% and 6% of isolates and were found in 17 and 15 locations, respectively. Two other types (types 42 and 47) accounted for 4% of the isolates and were found in 11 countries. The remaining isolates (n=1,517) were scattered into 235 types. Figure 1B shows the relative sizes of 259 shared types; 109 shared types (42%) contained only two patients each and 38 shared types contained only three patients each. Inversely, 24 shared types containing >20 patients totaled 1,804 (65%) isolates. Finally, the distribution of "unique" versus "ubiquitous" shared types (reported in one location versus found in two or more locations) is shown in Figure 1C; 122 (47%) shared types were reported from a single location, 69 (26%) were from two locations, and 25 (10%) were from three locations. Inversely, the most ubiquitous types, in increasing order of distribution, were 33 and 37, 20, 52, 42, 50, and 53. Thus, most M. tuberculosis shared types contained a low number of patient isolates and were confined geographically, whereas a minority contained a high number of patient isolates and were highly disseminated. The finding of identical spoligotypes in distant countries may be explained either by recent or past transmission events or by phylogenetic convergence. However, the evolution of the DR locus relies on at least three independent mechanisms, namely, homologous recombination (13), replication slippage (14,15), and insertion sequence-mediated transposition (16-19), which does not favor a fortuitous convergence.
Geographic Distribution of Shared Types in the Database
Analysis of geographic distribution of the shared types (see Figure A1) permitted us to split our collection into two broad categories: those reported in a single area (n=122, Table 2) and those reported in two or more areas (n=137). In the latter category, matching analysis for 69 spoligotypes found in four broad geographic areas, namely, Africa, the Americas (North, Central and Caribbean, and South America), Europe, and Asia (Middle East, and Far East Asia), is shown in Table 3. Contrary to ubiquitous spoligotypes such as type 1, 53, and 50, which have been found in all regions, this is an attempt to define potential inter-regional and inter-continental flow of M. tuberculosis isolates so far confined to limited geographic areas. The most frequent matches were found for clusters in European countries (n=17), followed by Europe and North America (n=8), Europe and Central America and the Caribbean (n=5), and Europe and South America (n=4) (Table 3). These matches may underline both recent transcontinental transmission events and the history of TB spread in the New World through European settlers.
A total of 25 shared types were reported in three countries. Among these, 8 types were exclusively found either in Europe (types 10,22,161) or the Americas (types 5,67,70,93,130); 10 types were shared between two European countries and a country of another region (types 35,49,59,86,115,118,136,138,139,150); 5 types were shared between two countries of the Americas with a country in Europe (types 92,119,168,185,190); 1 type was shared between a European country and two African countries (type 125); and 1 type was shared between Asia, Europe, and the USA (type 124). Finally, 15 types were found in four countries; 1 type (type 41) was exclusively found in Europe and may be specific for this continent. Fourteen other types were distributed as follows: Europe+Americas, 8 types (types 3,7,19,31,40,51,137,152); Europe+Africa, 1 type (type 21); Europe + Asia + Americas, 3 types (type 8,89,167); Europe + Americas + Africa, 1 type (type 64); and Europe + Africa + Asia, 1 type (type 126). Finally, 28 types were reported in five or more countries, suggesting that these types are widespread and may constitute the ubiquitous types such as the Beijing type (type 1 in our database) or the Haarlem type (type 47). The only exception in this category was type 17, which was found in six countries in the Americas and may be specific for this region. Future population studies should focus on these ubiquitous types to better define their relative prevalence in each country.
Biogeographic Analysis of European Versus American Spoligotypes
Several possible scenarios could account for the introduction and spread of TB in the Americas; however, documented contact with Europeans is considered too recent to account for the widespread distribution of the disease by AD 1000 (20). One hypothesis suggests that TB may have penetrated the Americas through human migration from Asia via the Bering Strait (21). Another scenario suggests TB's initial introduction as a zoonosis that became an anthropozoonosis after cattle were domesticated (20,21). In this context, of the 259 shared types in our database, 59 were exclusively reported in the Americas, whereas 50 were found only in Europe (Table 2). This biogeographic dichotomy may signal the specific history of the disease in each continent. As enough data were present for the USA and Europe (2,418 [73%] isolates), a statistical analysis of distribution of shared types found in those two areas was performed. 1 Of 45 shared types in this category, results showed that differences in the distribution of certain shared types (1,19,20,25,26,37,44,48,50,52,53,118,137) between the USA and Europe were highly significant, and sampling bias could not explain the differences observed (Table 4). On the other hand, the differences observed in the distribution of shared types 2,8, 33,34,47,58,62,92,138, and 139 between the USA and Europe were not statistically significant, and in this case sampling bias could not be fully excluded for the differences observed. Finally, our database described 58 isolates of the shared type 42 that were present in 11 countries (a ubiquitous type), but not a single isolate of type 42 was present among the 1,283 isolates from Texas (12).
Use of Database for Epidemiologic Studies
Essentially working in a Caribbean setting for last 6 years with systematic typing of all M. tuberculosis isolates from Guadeloupe, Martinique, and French Guiana, we initially focused on spoligotypes that may be specific to our region. Of 259 shared types, 85 types were present in the Caribbean. Of these, 69 were common to the Caribbean and the rest of the world, and 16 were reported only from the Caribbean (types 5,12,13,14,15,30,63,66,68,72,76,77,94,96,103,259). Although TB has a penchant to be latent for years or decades, because of an exhaustive (nearly 100%) recruitment of isolates from the French Caribbean for last 6 years, finding a previously unreported spoligotype in our region may constitute indirect evidence for a newly imported case of TB in most instances, particularly if an epidemiologic investigation does not suggest reactivation of old disease.
As far as global epidemiologic studies are concerned, this database also emphasizes the existence of highly prevalent families of M. tuberculosis isolates, e.g., the Beijing type, which represents a diverse collection of clones including the notorious multidrug-resistant strain W and other W-like drug-sensitive isolates (5,22). Studies focusing on M. tuberculosis isolates from developing countries, where TB is highly prevalent, would improve understanding of the worldwide circulation of tubercle bacilli and provide insights into their epidemiology, phylogeny, and virulence.
Phylogenetic Reconstruction of M. tuberculosis
Figure 2
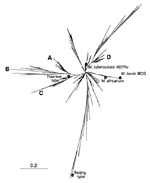
Figure 2. . Phylogenetic tree of shared types of Mycobacterium tuberculosis constructed by pairwise comparison of patterns using the "1-Jaccard" index and the neighbor-joining algorithm. Approximately 15 branches may be visualized at an...
Figure 3
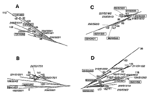
For phylogenetic analysis (23), a neighbor-joining tree was constructed by calculating the 1-Jaccard Index (10,24). This tree (Figure 2) incorporates the data for 252 M. tuberculosis shared types instead of the 259 types described in the online database (types 253 to 253 were added recently after the completion of phylogentic analysis). At an arbitrary distance of 0.2, one may easily distinguish nearly 15 branches that may contain significant phylogenetic information, as seen below for four selected branches (A to D) by combining results using independent genetic markers (Figure 3). As shown in Figure 2 and Figure 3A, the homogeneous branch A (mainly present in Europe, West Africa, and South America) contains 20 types characterized by the absence of spacer 29 to 32 and 34. Such a family of isolates was recently described in Guinea-Bissau and also found to harbor a low copy number of IS6110 (25). Information concerning katG283-gyrA95 allele combination was available for 5 of these 20 types and showed that branch A belonged to the major genotypic group 1 as defined previously (26) and may represent an ancestral clone of M. tuberculosis isolates originating in Africa, Asia, or both (27; this work). For this branch, VNTR information was available for 3 of 20 types and showed a high exact tandem repeat (ETR)-A copy number (between 4 to 7; Figure 3A), which is common both for M. bovis and M. africanum (8,28).
Branch B shared a common root with branch A (Figure 2) but was clearly distinct from the population in branch A, an observation corroborated both by VNTR and katG283-gyrA95 allele types (Figure 3B). All the isolates in branches A and B were of the major genetic group 1, as defined (26), except for a single isolate of the major genetic group 2 in branch B (type 199); the significance of this observation is not clear. Branch C was composed of two subbranches, which are likely to be of different phylogenetic significance (Figure 3C); the upper part related to the Haarlem family, as previously defined (15), and was highly homogeneous upon VNTR typing (alleles 32333), whereas the lower part was quite heterogenous (alleles 42431, 31333, 44553).
Finally, branch D comprised a subfamily of the spoligotypes that all missed spacers 33-36 (Figure 3D). This branch, which contained 30 different shared types, was easily characterized by simultaneous absence of spacers 21-24 and 33-36, and constitutes a highly ramified but homogeneous family on the basis of its belonging to the major genetic group 2 of Sreevatsan et al. (26), and the presence of two copies of the ETR-A allele upon VNTR typing. Frequently, found in southern Europe and Central and South America, the ancestral type of this family (type 42) may have evolved by stepwise mutation to give, successively, types 20 and 17 (Figure 3D). This assumption is corroborated by the position of the respective types in the tree and their spoligotyping and VNTR patterns; type 42 (all spacers present except 21 to 24 and 33 to 36, VNTR 22433), type 20 (identical to type 42 plus a single missing spacer 3, VNTR identical to type 42), and type 17 (identical to type 20 plus a single missing spacer 13, VNTR 22321).
These results show that branches A and B are likely to be of an older evolutionary origin than branches C and D. Källenius et al. (25) hypothesized that branches A and B could find their evolutionary origin in West Africa, whereas branches C and D could be of European descent. However, since the global evolutionary rate of the DR locus may involve many independent mechanisms, this tree is likely to incorporate systematic yet unknown errors (6); therefore, a detailed analysis of the robustness of each potential phylogenetic link is under investigation.
We have presented an update of a database of M. tuberculosis spoligotypes with a detailed description of 259 shared types. This database may help to address major aspects linked to recent mycobacterial reemergence, evolutionary history, and future epidemiologic studies. Our results demonstrate that a few major families of conserved spoligotypes are well distributed throughout the world, whereas others are specific for certain geographic regions. Thus, the current epidemiologic picture of TB appears to be based both on the persistence of ancestral clones of M. tuberculosis as well as those emerging more recently, e.g., the Beijing type (type 1 in our database), which also includes the MDR strain W from New York City. A future correlation between genotyping and resistance data and the respective prevalence of various clones region by region may provide more insight into the global circulation of TB and help establish priorities in TB control programs. For example, because we have typed all M. tuberculosis clinical isolates in our insular setting for last 6 years, introduction of a previously unreported clone in Guadeloupe may be detected and, when placed in epidemiologic context, may either be classified as a newly imported case of TB or as a reactivation. Simultaneously, an epidemiologic investigation around the case is immediately initiated by local health authorities. A comparison of the newly imported clone with those in the database sometimes suggests a probable link to a specific community or, alternatively, regional, national, or intercontinental importation of the disease.
Concerning the global phylogeny of M. tuberculosis, the pairwise comparison of the 252 shared types by calculation of the 1-Jaccard index and the neighbor-joining algorithm underscored phylogenetic relationships between some of the families of spoligotypes described. Four major families of spoligotypes (branches A-D) were discussed in detail, and the results were corroborated by VNTR and katG--gyrA polymorphism data, which support the robustness of the branchings proposed. Nevertheless, a detailed and more exhaustive analysis of evolutionary and historical spreading of the different families of tubercle bacilli is a long-term goal requiring a never-ending compilation of data. Ideally, this database could be expanded to incorporate detailed M. bovis and M. africanum results so as to infer the global phylogeny of all members of the M. tuberculosis complex.
It has been suggested that the evolutionary rate of M. tuberculosis may be strain dependent (29). In this context, our investigation also pointed out a previously unnoticed link between spoligotypes and the katG--gyrA polymorphism (Figure 3), e.g., the isolates in the spoligotyping-defined branch A belonged to the major genetic group 1 of Sreevatsan et al. (26), whereas those in branch D belonged to the major genetic group 2. Since the isolates in these branches came from diverse geographic areas, we suggest that the pace of the molecular clock of the DR locus might be much slower than that of other markers, such as IS6110. This assumption is supported by a recent study on the evolutionary origin of the DR locus of M. tuberculosis (19). Finally, by comparing observations with outcomes of a stepwise mutation model, the insertion sequences of the tubercle bacilli are far from equilibrium; indeed, transposition parameters appear to have a much stronger effect on IS6110 copy number distribution than epidemic parameters and have a direct action on bacterial diversity of the M. tuberculosis complex (30). New studies are needed to clarify the complex relationships between epidemic parameters, selection factors, and genomic evolutionary mechanisms of the tubercle bacilli.
Dr. Sola is a senior scientist at the Pasteur Institute and has been working at the Institut Pasteur de Guadeloupe for the last 6 years. His current research interest focuses on molecular population genetics of tubercle bacilli for public health and academic purposes.
Acknowledgments
We thank Olga Narvskaya, Sofia Samper, Carlos Martin, Bernard Carbonnelle, and Jéroôme Maïsetti for permission to use their unpublished results in our database. We are grateful to Prasitt Palittapongarnpim and Richard Frothingham for providing some of the DNAs used for spoligotyping and permission to use their unpublished results in our database.
This work was supported through grants by the Délégation Générale au Réseau International des Instituts Pasteur et Instituts Associés, Institut Pasteur, Paris, and Fondation Française Raoul Follereau, Paris, France.
References
- World Health Organization. Global tuberculosis control. WHO report 1999. Geneva: The Organization; 1999.
- Slutkin G. Global AIDS 1981-1999: the response. Int J Tuberc Lung Dis. 2000;4:S24–33.PubMedGoogle Scholar
- Snider DE, Castro KG. The global threat of drug resistant tuberculosis. N Engl J Med. 1998;338:1689–90. DOIPubMedGoogle Scholar
- Long R, Nobert E, Chomyc S, van Embden J, McNamee C, Duran RR, Transcontinental spread of multidrug-resistant Mycobacterium bovis. Am J Respir Crit Care Med. 1999;159:2014–7.PubMedGoogle Scholar
- Bifani PJ, Mathema B, Liu Z, Moghazeh SL, Shopsin B, Tempalski B, Identification of a W variant outbreak of Mycobacterium tuberculosis via population-based molecular epidemiology. JAMA. 1999;282:2321–7. DOIPubMedGoogle Scholar
- Sola C, Devallois A, Horgen L, Maïsetti J, Filliol I, Legrand E, Tuberculosis in the Carribean: using spacer oligonucleotide typing to understand strain origin and transmission. Emerg Infect Dis. 1999;5:404–14. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.PubMedGoogle Scholar
- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144:1189–96. DOIPubMedGoogle Scholar
- Grimont PAD. TAXOTRON instruction manual. Paris: Taxolab, Institut Pasteur; 1996.
- Jaccard P. Nouvelles recherches sur la distribution florale. Bull Soc Vaud Sci Nat. 1908;44:223–70.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.PubMedGoogle Scholar
- Soini H, Pan X, Amin A, Graviss EA, Siddiqui A, Musser JM. Characterization of Mycobacterium tuberculosis isolates from patients in Houston, Texas, by spoligotyping. J Clin Microbiol. 2000;38:669–76.PubMedGoogle Scholar
- Groenen PMA, Bunschoten AE, van Soolingen D, van Embden JDA. Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Mol Microbiol. 1993;10:1057–65. DOIPubMedGoogle Scholar
- Hancock JM. The contribution of slippage-like processes to genome evolution. J Mol Evol. 1995;41:1038–47. DOIPubMedGoogle Scholar
- Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PWM, Martin C, Comparison of methods based on different molecular epidemiologial markers for typing of Mycobacterium tuberculosis strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol. 1999;37:2607–18.PubMedGoogle Scholar
- Fang Z, Doig C, Kenna DT, Smittipat N, Palittapongarnpim P, Watt B, IS6110-mediated deletions of wild-type chromosomes of Mycobacterium tuberculosis. J Bacteriol. 1999;181:1014–20.PubMedGoogle Scholar
- Fang Z, Morrison N, Watt B, Doig C, Forbes KJ. IS6110 transposition and evolutionary scenario of the direct repeat locus in a group of closely related Mycobacterium tuberculosis strains. J Bacteriol. 1998;180:2102–9.PubMedGoogle Scholar
- Filliol I, Sola C, Rastogi N. Detection of a previously unamplified spacer within the DR locus of Mycobacterium tuberculosis: Epidemiological implications. J Clin Microbiol. 2000;38:1231–4.PubMedGoogle Scholar
- van Embden JDA, van Gorkom T, Kremer K, Jansen R, Van der Zeijst BAM, Schouls LM. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J Bacteriol. 2000;182:2393–01. DOIPubMedGoogle Scholar
- Buikstra JE. Paleoepidemiology of tuberculosis in the Americas. In: Palfi G, Dutour O, Deak J, Hutas I, editors: Tuberculosis: past and present. Szeged, Hungary: Golden Book Publisher Ltd.; 1999. p. 479-94.
- Ortner DJ. Paleopathology: implications for the history and evolution of tuberculosis. In: Palfi G, Dutour O, Deak J, Hutas I, editors. Tuberculosis: past and present. Szeged, Hungary: Golden Book Publisher Ltd; 1999. p. 255-61.
- Kurepina NE, Sreevatsan S, Plikaytis BB, Bifani PB, Connell ND, Donneelly RJ, Characterization of the phylogenetic distribution and chromosomal insertion sites of five IS6110 elements in Mycobacterium tuberculosis: non random integration in the dnaA-dnaN region. Tuber Lung Dis. 1998;79:31–42. DOIPubMedGoogle Scholar
- Nei M. Phylogenetic analysis in molecular evolutionary genetics. Annu Rev Genet. 1996;30:371–403. DOIPubMedGoogle Scholar
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.PubMedGoogle Scholar
- Källenius G, Koivula T, Ghebremichael S, Hoffner SE, Norberg R, Svensson E, Evolution and clonal traits of Mycobacterium tuberculosis in Guinea-Bissau. J Clin Microbiol. 1999;37:3872–8.PubMedGoogle Scholar
- Sreevatsan S, Pan X, Stockbauer K, Connell N, Kreiswirth B, Whittam T, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;97:9869–74. DOIPubMedGoogle Scholar
- Castets M, Boisvert H, Grumbach F, Brunel M, Rist N. Les bacilles tuberculeux de type africain: note préliminaire. Rev Tuberc Pneumol (Paris). 1968;32:179–84.PubMedGoogle Scholar
- Frothingham R, Strickland PL, Bretzel G, Ramaswamy S, Musser JM, Williams DL. Phenotypic and genotypic characterization of Mycobacterium africanum isolates from West Africa. J Clin Microbiol. 1999;37:1921–6.PubMedGoogle Scholar
- Warren GM, Richardson M, Sampson S, Bourn W, van der Spuy G, Hide W, RFLP analysis of M. tuberculosis demonstrates strain-dependent evolution. Int J Tuberc Lung Dis. 1999;3(Suppl. I):S38.
- Tanaka MM, Small PM, Salamon H, Feldman MW. The dynamics of repeated elements: applications to the epidemiology of tuberculosis. Proc Natl Acad Sci U S A. 2000;97:3532–7. DOIPubMedGoogle Scholar
- Bauer J, Andersen AB, Kremer K, Miörner H. Usefulness of spoligotyping to discriminate IS6110 low-copy-number Mycobacterium tuberculosis complex strains cultures in Denmark. J Clin Microbiol. 1999;37:2602–6.PubMedGoogle Scholar
- Bonora S, Gutierrez MC, Perri GD, Brunello F, Allegranzi B, Ligozzi M, Comparative evaluation of Ligation-mediated PCR and spoligotyping as screening methods for genotyping of Mycobacterium tuberculosis strains. J Clin Microbiol. 1999;37:3118–23.PubMedGoogle Scholar
- Diaz R, Kremer K, de Haas PEW, Gomez RI, Marrero A, Valdivia JA, Molecular epidemiology of tuberculosis in Cuba outside of Havana, July 1994-June 1995: utility of spoligotyping versus IS6110 restriction fragment length polymorphism. Int J Tuberc Lung Dis. 1998;2:743–50.PubMedGoogle Scholar
- Douglas JT, Qian L, Montoya JC, Sreevatsan S, Musser J, van Soolingen D, Detection of a novel family of tuberculosis isolates in the Philippines. 97th general meeting of the American Society for Microbiology. Washington: ASM Press; 1997. p.572.
- Escalante P, Ramaswamy S, Sanabria H, Soini H, Pan X, Valiente-Castillo O, Genotypic characterization of drug-resistant Mycobacterium tuberculosis isolates from Peru. Tuber Lung Dis. 1998;79:111–8. DOIPubMedGoogle Scholar
- Goguet dela Salmonière YO, Li HM, Torrea G, Bunschoten A, van Embden JDA, Gicquel B. Evaluation of spoligotyping in a study of the transmission of Mycobacterium tuberculosis. J Clin Microbiol. 1997;35:2210–4.PubMedGoogle Scholar
- Goyal M, Saunders NA, van Embden JDA, Young DB, Shaw RJ. Differentiation of Mycobacterium tuberculosis isolates by spoligotyping and IS6110 restriction fragment length polymorphism. J Clin Microbiol. 1997;35:647–51.PubMedGoogle Scholar
- Heyderman RS, Goyal M, Roberts P, Ushewokunze S, Zishou S, Marshall BG, Pulmonary tuberculosis in Harare, Zimbabwe: analysis by spoligotyping. Thorax. 1998;53:346–50. DOIPubMedGoogle Scholar
- Niang MN, Goguet de la Salmonière YO, Samb A, Hane AA, Cisse MF, Gicquel B, Characterization of M. tuberculosis strains from West-African patients by spoligotyping. Microbes Infect. 1999;1:1189–92. DOIPubMedGoogle Scholar
- Popa MI, Goguet de la Salmonière YO, Teodor I, Popa L, Stefan M, Banica D, Genomic profile of Romanian M. tuberculosis strains appreciated by spoligotyping. Roum Arch Microbiol Immunol. 1997;56:63–75.PubMedGoogle Scholar
- de C. Ramos M. Soini H, Roscanni GC, Jaques M, Villares MC, Musser JM. Extensive cross-contamination of specimens with Mycobacterium tuberculosis in a reference laboratory. J Clin Microbiol. 1999;37:916–9.PubMedGoogle Scholar
- Taylor GM, Goyal M, Legge AJ, Shaw RJ, Young D. Genotypic analysis of Mycobacterium tuberculosis from medieval human remains. Microbiology. 1999;145:899–904. DOIPubMedGoogle Scholar
- van der Zanden AG, Hoentgen AH, Heilmann FG, Weltvreden EF, Schouls LM, van Embden JD. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis complex in paraffin wax embedded tissues and in stained microscopic preparations. Mol Pathol. 1998;51:209–14. DOIPubMedGoogle Scholar
- van Soolingen D, Qian L, de Haas PEW, Douglas JT, Traore H, Portaels F, Predominance of a single genotype of Mycobacterium tuberculosis in countries of East Asia. J Clin Microbiol. 1995;33:3234–8.PubMedGoogle Scholar
Figures
Tables
Cite This Article1For this purpose, the independent sampling sizes for Europe and the USA were taken as n1 and n2, the number of individuals within a given shared-type "x" was k1 and k2, and in this case, the representativeness of the two samples was p1=k1/n1 and P2=k2/n2, respectively. To assess if the divergence observed between p1 and p2 was due to sampling bias or the existence of two distinct populations, the percentage of individuals (p0) harboring shared-type "x" in the population studied was estimated by the equation p0= k1+k2/n1+n2=n1p1+n2p2/n1+n2. The distribution of the percentage of shared-type "x" in the sample sizes n1 and n2 follows a normal distribution with a mean p0 and a standard deviation of 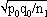 and
and 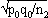 respectively, and the difference d=p1-p2 follows a normal distribution of mean p0-p0=0 and of variance σd2=σp12+σp22 = p0q0/n1+p0q0/n2 or σd2=p0q0 (1/n1+1/n2). The two samples being independent, the two variances were additive; the standard deviation σd=
respectively, and the difference d=p1-p2 follows a normal distribution of mean p0-p0=0 and of variance σd2=σp12+σp22 = p0q0/n1+p0q0/n2 or σd2=p0q0 (1/n1+1/n2). The two samples being independent, the two variances were additive; the standard deviation σd=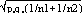 . If the absolute value of the quotient d/σd<2, the two samples were considered to belong to a same population (CI 95%) and the variation observed in the distribution of isolates for given shared types could be due to a sampling bias. Inversely, if d/σd>2, then the differences observed in the distribution of isolates for given shared types were statistically significant and not due to potential sample bias.
. If the absolute value of the quotient d/σd<2, the two samples were considered to belong to a same population (CI 95%) and the variation observed in the distribution of isolates for given shared types could be due to a sampling bias. Inversely, if d/σd>2, then the differences observed in the distribution of isolates for given shared types were statistically significant and not due to potential sample bias.
Table of Contents – Volume 7, Number 3—June 2001
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nalin Rastogi or Christophe Sola, Unité de la Tuberculose et des Mycobactéries, Institut Pasteur de Guadeloupe, Morne Jolivière, BP 484, 97165 Pointe-à-Pitre, Cedex, Guadeloupe; fax: 590-893880Nalin Rastogi or Christophe Sola, Unité de la Tuberculose et des Mycobactéries, Institut Pasteur de Guadeloupe, Morne Jolivière, BP 484, 97165 Pointe-à-Pitre, Cedex, Guadeloupe; fax: 590-893880
Top