Volume 15, Number 9—September 2009
Research
Distant Relatives of Severe Acute Respiratory Syndrome Coronavirus and Close Relatives of Human Coronavirus 229E in Bats, Ghana
Cite This Article
Citation for Media
Abstract
We tested 12 bat species in Ghana for coronavirus (CoV) RNA. The virus prevalence in insectivorous bats (n = 123) was 9.76%. CoV was not detected in 212 fecal samples from Eidolon helvum fruit bats. Leaf-nosed bats pertaining to Hipposideros ruber by morphology had group 1 and group 2 CoVs. Virus concentrations were <45,000 copies/100 mg of bat feces. The diversified group 1 CoV shared a common ancestor with the human common cold virus hCoV-229E but not with hCoV-NL63, disputing hypotheses of common human descent. The most recent common ancestor of hCoV-229E and GhanaBt-CoVGrp1 existed in ≈1686–1800
Coronaviruses (CoVs) (order Nidovirales, family Coronaviridae, genus Coronavirus) are enveloped viruses with plus-stranded RNA genomes of 26–32 kb, the largest contiguous RNA genomes in nature (1). They are classified into 3 groups, which contain viruses pathogenic for mammals (groups 1 and 2) and poultry (group 3) (1). Human CoVs (hCoVs)-229E, -NL63, -OC43, and -HKU1 are endemic worldwide and cause mainly respiratory infections in children and adults. The severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) is a novel zoonotic coronavirus that caused an international epidemic in 2002–2003. Fortunately, efficient public health management interrupted this epidemic (2). Studies conducted in China in the aftermath of the SARS epidemic have identified CoVs in bats (Chiroptera) and implicated this speciose mammalian order as the most likely reservoir of all known coronaviruses (3–7). Among the most urgent concerns prompted by the SARS epidemic is the likelihood of similar future events. Thus, it seems highly relevant to study the ecology of bat CoVs in terms of diversity, host restriction, virus prevalence, risk of exposure, and the circumstances of past host transition events.
The genetic diversity of bat-borne CoVs is currently unclear. Preliminary data suggest that CoVs may be adapted in a stricter sense to a specific host species rather than to specific regions (5,6,8–12). A variety of pathogenic CoVs occur in other mammals or poultry. However, the genetic range within these animals is considerably less than that observed in even single bat species or subfamilies (7,8).
Estimates indicate that there are >100 bat species in sub-Sahran Africa. This finding is in contrast to ≈50 species in the entire Western Palaearctic region (Europe, Middle East, North Africa) (13,14). African bats have been shown to harbor pathogens that are occasionally transmitted to humans. This transmission may result in severe disease outbreaks, e.g., Ebola and Marburg viruses (15). Because bats are a part of the human diet in wide areas of Africa (16), it appears highly relevant to study CoVs in African bats.
We have demonstrated by serologic studies that African bats have antibodies against CoVs (10). Antibodies reactive with SARS-CoV antigen were detected in 47 (6.7%) of 705 bat serum specimens from 26 species (10). Recently, Tong et al. detected sequences of CoVs in bats from Kenya (17). We describe the results of studies on bats in Ghana obtained by using noninvasive sampling of frugivorous and insectivorous bats at 2 caves, a lake habitat of diverse insectivorous bats, and a large urban roosting site of frugivorous bats. Bayesian inference of diversification dates gave implications on the recency of the introduction of hCoV-229E into the human population, irrespective of its original source.
Capturing and Sampling
Figure 1
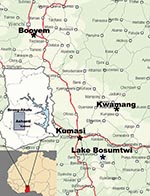
Figure 1. Location of Kwamang caves near the village of Kwamang, (6°58′N, 1°16′W), 50 km northeast of Kumasi, Ashanti region, Ghana. Booyem caves A (7°43′24.9′′N, 1°59′16.5′W) and B (7°43′25.7′′N, 1°59′33.5′′W) are located near...
In the locations identified in Figure 1, mist netting and sampling were conducted as described (11). In Kumasi Zoo, fecal samples were collected with plastic foil under trees occupied by Eidolon helvum bats (estimated colony size 300,000). For all capturing and sampling, permission was obtained from the Wildlife Division of the Ministry of Lands, Forestry, and Mines in Ghana. Research samples were exported under a state agreement between the Republic of Ghana and the Federal Republic of Germany, represented by the City of Hamburg. Additional export permission was obtained from the Veterinary Services of the Ghana Ministry of Food and Agriculture.
Processing and Analysis of Samples
Samples (1–4 fecal pellets or swabs suspended in RNA stabilization solution [RNAlater Tissue Collection; Applied Biosystems, Foster City, CA, USA]) were tested at the Kumasi Centre for Collaborative Research in Tropical Medicine as described (11,18). After initial sequencing, specific primers were designed for each group of CoV found. Nested reverse transcription–PCR (RT-PCR) primer sets used for sequencing of longer fragments of representative viruses are available upon request. The following sequences were derived from this study and were submitted to GenBank under the listed accession numbers: RNA-dependent RNA polymerase (RdRp) sequences: BtCoV/Hip/GhanaBoo/348/2008, FJ710043; BtCoV/Hip/GhanaBoo/344/2008, FJ710044; BtCoV/Hip/GhanaKwam/8/2008, FJ710045; BtCoV/Hip/GhanaKwam/19/2008, FJ710046; BtCoV/Hip/GhanaKwam/20/2008, FJ710047; BtCoV/Hip/GhanaKwam/13/2008, FJ710048; BtCoV/Hip/GhanaKwam/31/2008, FJ710049; BtCoV/Hip/GhanaKwam/27/2008, FJ710050; BtCoV/Hip/GhanaKwam/26/2008, FJ710051; BtCoV/Hip/GhanaKwam/24/2008, FJ710052; BtCoV/Hip/GhanaKwam/10/2008, FJ710053; BtCoV/Hip/GhanaKwam/22/2008, FJ710054/nucleocapsid sequences; BtCoV/Hip.sp/GhanaBoo/344/2008, FJ710055; BtCoV/Hip.sp/GhanaKwam/19/2008, FJ710056.
Phylogenetic Analysis
Nucleic acid alignments were conducted based on amino acid code by using the ClustalW algorithm (www.ebi.ac.uk/clustalw) in the Molecular Evolutionary Genetics Analysis version 4.0 software package (www.megasoftware.net) (19). Two gap-free nucleotide alignments (817 bp and 1,221 bp) were generated. Tree topologies were determined on both datasets by using MrBayes version 3.1 (20). The analysis used a general time reversible (GTR) substitution model with 6 rate categories to approximate a gamma-shaped rate distribution across sites and an invariant site assumption (GTR + Γ6 + I). Markov chain Monte Carlo (MCMC) chains of 107 iterations were sampled every 500 generations, resulting in 20,000 sampled trees. Two Metropolis-coupled chains (1 cold and 3 heated chains each) were run in parallel, compared, and pooled. Convergence of chains was confirmed by the potential scale reduction factor statistic in MrBayes (21) and by visual inspection of each cold chain using the TRACER program (22). Phylogenetic dating was conducted by using Bayesian evolutionary analysis sampling trees (BEAST) (22). Chain lengths in BEAST were at least 20,000,000 generations with sampling every 500 generations. Convergence of the model was checked visually and by the effective sample size statistic with TRACER.
Virus Detection
During February 2008, bats were sampled in the described locations around Kumasi, Ghana. Initially, 7 fecal samples tested positive by pan-CoV PCR. Products (440 bp, RdRp gene) were sequenced and aligned with prototype CoV. Neighbor-joining phylogenies indicated 2 distinct groups of sequences that belonged to CoV group 1 (n = 4) and group 2 (n = 3), respectively. Specific primer pairs for the group 1 and group 2 sequences were designed and applied again to all samples. Five additional viruses were found, resulting in a total CoV prevalence of 9.76% in insect-eating bats (n = 123). No virus was found in any oral swab. All virus findings in fecal samples are listed by capture site in Table 1.
Figure 2

Figure 2. Two morphotypes of Hipposideros caffer ruber bats held by one of the authors (F.G.-R.), who was wearing a leather glove. Photograph courtesy of Antje Seebens.
Notably, all CoV findings were in insect-eating leaf-nosed bats of the genus Hipposideros. Within the genus, the species H. abae could be discriminated unambiguously by morphology (Table 1). The remaining Hipposideros species were assigned to the complex of forms related to currently recognized species H. caffer and H. ruber. Because 2 morphotypes were present (Figure 2), the mitochondrial cytochrome b gene was sequenced as described (23). Both morphotypes belonged to phylogenetic lineages distinct from H. caffer and possibly represented 2 distinct species (P. Vallo, personal ongoing investigation). Both are collectively referred to as H. caffer (cf.) ruber in this study. A fraction of 15.4% of H. cf. ruber specimens yielded CoV, without a difference between sexes (14%/19%, n = 57/21 [M/F], respectively). Only adult males and nonlactating adult females, but no lactating females, juveniles, and subadults of H. cf. ruber were encountered.
Virus Concentrations
To estimate the quantity of CoV genomes in bat feces, we did end-point dilution experiments with the nested pan-CoV RT-PCR (18). The previously determined sensitivity limit of the PCR assay was 5–45 copies/PCR (18). In the assay, the equivalent of 1 mg feces was tested per PCR tube (100 mg feces collected, 1:10 dilution extracted, 1:10 dilution tested). The highest dilution factor that still yielded an amplification signal in any of the samples was 1:10, which suggested a maximal concentration of 50 to 450 CoV RNA copies/mg of feces.
Virus Classification
Group 1 CoV
Figure 3

Figure 3. A) Phylogeny of coronaviruses (CoVs) in the RNA-dependent RNA polymerase gene (RdRp, 817-bp fragment) with root point dates derived from Bayesian inference under a relaxed lognormal molecular clock assumption with a...
In H. cf. ruber bats in the Kwamang and Booyem caves, a diverse group 1 CoV was found. Further analysis was complicated by the low RNA content in samples. Based on alignments of prototype group 1 viruses, 5 different nested RT-PCRs were designed and the RdRp fragment could finally be extended by 441 bp to the 5′ end, providing an 817-nt fragment for phylogenetic analysis. All methods of phylogenetic inference placed this virus next to a common ancestor with human coronavirus 229E, which circulates worldwide in humans (Figure 3). Bootstrap support of the hCoV-229E/GhanaBt-CoVGrpI root point in neighbor-joining analysis was 100%. The corresponding Bayesian posterior probability was 1.0. The most closely related member of the GhanaBt-CoVGrp1 clade shared 91.90% nucleotide identity with hCoV-229E in the analyzed fragment. The most distant member was 86.50% identical. The next phylogenetic neighbor, the human CoV hCoV-NL63, was only 74.70%–78.60% identical in the analyzed fragment.
Group 2 CoV
With the pan-CoV screening assay, a group 2 CoV was initially found in the Kwamang cave. Sequences from 3 bats were identical. The secondary group-specific PCR identified 4 additional samples of this virus, 1 of them from Booyem Cave B and the remaining from Kwamang. Nucleotide identity among these sequences was 97.2%–100%. Phylogenetic analysis with different methods of inference (neighbor-joining nucleotide-based, neighbor-joining amino acid–based, Bayesian) yielded variable tree topologies suggesting basal associations with either the 2a, 2d, or 2b subgroups (data not shown) (24). Based on alignments of prototype group II viruses, 8 additional nested RT-PCR primer sets were designed and 2 of the samples could be amplified. Sequences could be extended 520 bp upstream and 383 bp downstream of the initial fragments, yielding 1,221-bp fragments for phylogenetic analysis. Bayesian phylogenetic inference with different substitution models and parallel analysis using Metropolis coupling now placed the virus reliably next to a common ancestor with the 2b group of CoV (SARS-like viruses, Figure 3). The Bayesian posterior probability of the CoV 2b/GhanaBt-CoVGrp2 clade being monoyphletic was 1.0. A maximum of 72.2% nucleotide identity was shared with SARS CoV.
Molecular Dating
Reliable isolation dates were researched in the literature for each employed virus. Because a reliable molecular clock dating existed for the most recent common ancestor (MRCA) of the hCoV-OC43/bovine CoV pair (25), this date was set as a normal-distributed probabilistic prior within the published ranges (25) for calibration of all analyses. A first analysis was conducted on the 1,225-bp dataset that did not include the novel GhanaBt-CoVGrp1. All virus sequences were assumed to be contemporary. Phylogeny was inferred using a GTR + Γ4 + I model. The resulting MRCA date of the CoV2b (SARS-like)/GhanaBt-CoVGrp2 clade was 260
Because it has been suggested that codon-based evolutionary models may be preferred for Bayesian phylogenetic inference from protein-coding datasets (27), analyses on the 817-bp dataset were repeated by using the SRD06 substitution model in BEAST. This analysis did not yield a different substitution rate, but resulted in older resulting MRCA dates (Table 2). A Bayes factor test conducted in TRACER yielded a strong estimate of superiority of the codon-based model over the GTR + Γ4 + I model (log10 Bayes factor 139 [20 is highly significant]). To further optimize the prediction of MRCA dates, the constant population size assumption used in all analyses was exchanged against expansion growth or exponential growth assumptions. Both assumptions were predicted to fit the data better than the constant size model (Bayes factors 13.5 and 13.9). There was no difference between the expansion and exponential models (Bayes factor 0.34 in favor of expansion). The MRCA date of hCoV-229E and the GhanaBt-CoVGrp1 was 1686 (expansion) or 1800 (exponential growth). Table 2 summarizes the results. Figure 3 shows a dated phylogeny of coronaviruses with MRCAs according to the 2 last mentioned analyses.
Recombination
To determine whether CoV recombination might play a role in the studied virus population, the structural nucleocapsid gene was amplified using 8 nested RT-PCR primer sets that had been designed on alignments of all available CoV group 1 nucleocapsid sequences. Using a similar approach, we also tested the same samples for CoV group 2 nucleocapsid sequences. Only group 1 RT-PCRs yielded fragments. These fragments could be combined into contiguous 1,030-nt sequences for Bt-CoV GhanaKwam 19 and 1,176 nt for Bt-CoV GhanaBoo 344. As shown in Figure 3, panel B, the resulting phylogenetic placement was exactly matching that of the RdRp fragments, giving no evidence of recombination between the RdRp region located in the middle of the genome and the nucleocapsid gene located at the extreme downstream end. Sequencing of the nucleocapsid gene of the GhanaBt-CoVGrp2 was not successful when we used 15 nested RT-PCRs designed on alignments of all available CoV 2b nucleocapsid sequences. Amplification with above mentioned nested RT-PCRs for CoV group 1 was also unsuccessful.
In the aftermath of the SARS epidemic, bats have been identified as carriers of CoV in China (3–7). Furthermore, in addition to our earlier finding of antibodies against CoVs in various African bats (10), we have confirmed the presence of CoV in bats of Ghana. Together with recent data from Germany, North America, Trinidad, and Kenya (11,12,28), these findings suggest that the association of CoV with bats is a worldwide phenomenon. The prevalence of CoV in insect-eating bats (9.76%) matched our previous findings in Germany. However, in that study we sampled during the breeding season and showed that CoVs are most likely amplified in maternity roosts (11). The composition of the catch in this study (no lactating females, no young bats) suggests sampling outside the breeding season and may not be directly comparable. Future studies relating to risks of exposure should address whether virus prevalence may change over time.
The risk of exposure was also addressed by investigations of virus concentration. Several groups have shown that CoVs are almost exclusively detected in bat feces and not, as hypothesized earlier, in saliva (3,4,28,29). Surprisingly, little virus was found in all fecal samples tested in our study. We estimated the RNA concentration per full sample (100 mg feces = 2–4 fecal pellets) to be only up to 4.5 × 104 RNA copies. Human pathogenic viruses transmitted by the fecal-oral route generate much higher virus concentrations in stool, up to ≈1012 RNA copies/mg, e.g., for different picornaviruses (30–32). Based on these data it would be difficult to postulate that humans can acquire CoV from bat feces. However, studies in other locations and at different times are needed to address virus concentration in bat droppings in more detail. Because virus in this study was only observed in insectivorous bats and not in frugivorous bats, future studies should investigate whether insects might constitute a source of CoV infection for bats.
To achieve a direct prediction of the potential of bat CoVs to infect human cells, it would be highly relevant to conduct virus isolation studies on bat feces. However, in our study we sampled no more than 100 mg of feces per bat. All samples had to be collected in RNAlater solution (0.5 mL) (Applied Biosystems) for reasons of storage and transportation. Although it has been suggested that RNAlater solution may preserve virus infectivity (33,34), our observations showed that the solution has to be diluted at least 1:20 in cell culture medium to avoid cytotoxicity (data not shown). Because of the low virus RNA concentrations observed, we did not attempt to isolate the virus. However, the absence of successful virus isolation from bat feces in previous studies (3–6,8,11,12) may not reflect the incapability of bat CoV to infect human cells. Recently, a synthetic bat CoV complemented with an appropriate spike protein has shown potential to infect human cells (35).
Reconstruction of phylogenetic and temporal relationships between bat CoV and other mammalian CoV is another way to obtain information on their zoonotic potential. Unfortunately, for CoV long sequence fragments must be analyzed before valid phylogenies can be inferred from the conserved nonstructural genome portion (28,36). Because of the low concentration of RNA in bat samples, generation of long sequences from novel bat CoV is tedious and technically demanding, which may be why some published phylogenies of bat CoV are based on short datasets, making it difficult to use these data for reference. For molecular clock dating, we have therefore relied on reference viruses mainly from other mammals that covered our 1,221-bp fragment in the conserved RdRp region. We assumed that the RdRp would be under less selective pressure than the structural genes and other nonstructural genes, and therefore could be used to infer nucleotide substitution rates over distantly related CoVs (7,25,26,36–38). We have confirmed all tree topologies using alternative methods of phylogenetic inference, including an MCMC algorithm implemented in MrBayes that eliminates artifacts contributed by fixation of MCMC chains in suboptimal prosterior probability maxima (20). Calibration was conducted on reliable isolation dates of prototype and novel bat CoV from the literature, as well as on the MRCA of the hCoV-OC43/Bovine CoV clade. For dating of only this specific CoV clade, a wide range of dated virus isolates has been available that covered as much as 34% (1965–2004) of the projected time of virus evolution from root to tip (1890–2004) (25). A probabilistic calibration prior was used, which is favorable for dating in combination with relaxed molecular clock assumptions (39). The determined mean substitution rates were in good concordance with earlier studies on non-bat–CoV that used maximum likelihood-based methods in addition to Bayesian inference (25,26,38,40).
Although the exponential growth prior on the virus population seemed equivalent with an expansion growth model by the Bayes factor test and produced highly compatible MRCAs, the exponential model produced a better match with the previously determined MRCA of the HCoV-NL63/HCoV-229E pair (26). Because Pyrc et al. generated these data by 3 alternative approaches (Bayesian, serial unweighted pair group method with arithmetic mean, maximum likelihood [26]), we used their MRCA to validate our results, and consequently prefer the MRCA dating from the exponential growth population model (as presented in Figure 3 in plain type). One earlier study on bat and non-bat CoV suggested a much faster evolutionary rate for CoV than other studies (7). As Vijaykrishna et al. pointed out, their results were associated with large confidence intervals caused by the lack of available data on Bt CoV at the time the study was conducted (7). The increase of available sequence data now enables a better account of CoV evolutionary history.
All CoVs in our study were found in members of the genus Hipposideros (family Hipposideridae). The genus Rhinolophus from the sister family Rhinolophidae was found to host SARS-like viruses in several studies in China. One of our Hipposideros CoVs was in a basal phylogenetic relationship with the SARS-like clade (group 2b); their most recent common ancestors date back to ≈400
Another result that should be integrated with earlier findings is the surprisingly recent date of the MRCA of the novel Grp1 Bt CoV and the human common cold virus hCoV-229E. Further to the proven recent host switching of SARS CoV, Vijgen et al. have suggested that hCoV-OC43 entered the human population ≈120 years ago, causing a pandemic (25). This virus was most likely acquired by humans from domestic cattle. Results of our study show that it is not unlikely that hCoV-229E, which today is circulating worldwide in humans, resulted from a host switching event not more than 208–322 years ago. However, as with molecular clock dating of viruses, associated confidence limits should not be overlooked.
Because H. cf. ruber bats are found only in sub-Saharan Africa and are not migratory (23), it would be relevant to know how tightly the associated CoV is restricted to its host. Despite the statistical limitations of our rather small sample size, the absence of CoV in bats of the closely related species H. abae that were tested in our study in two different caves speaks in favour of tight host restriction. Another supportive argument is the absence of CoV in C. afra, a bat species sampled in sufficient numbers at the Booyem cave. This cave was coinhabited by CoV-positive H. cf. ruber bats. If tight host restriction to nonmigratory H. cf. ruber bats existed, this would indicate an origin of hCoV-229E within the geographic range of its host, i.e., the rainforest belt and the wet forested savannahs of sub-Saharan Africa (23). Unfortunately, it will be difficult to reconstruct whether the projected host transition event might have been associated with human epidemic disease.
Dr Pfefferle is a physician and molecular biologist at the Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany. Her research focuses on coronavirus reverse genetics and molecular aspects of coronaviral host switching.
Acknowledgments
We are grateful to the voluntary staff at Noctalis for support.
This study was supported by the German Ministry of Education and Research (Project Code Ökologie und Pathogenese von SARS), the European Commission (contracts SSPE-CT-2005-022639 and FP7-INFRASTRUCTURES-2008-No 228292), and the Bundesamt für Bevölkerungsschutz und Katastrophenhilfe (contract BBK-F-440-00-1).
References
- Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629–33.PubMedGoogle Scholar
- Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–5. DOIPubMedGoogle Scholar
- Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–9. DOIPubMedGoogle Scholar
- Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–9. DOIPubMedGoogle Scholar
- Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80:7481–90. DOIPubMedGoogle Scholar
- Vijaykrishna D, Smith GJ, Zhang JX, Peiris JS, Chen H, Guan Y. Evolutionary insights into the ecology of coronaviruses. J Virol. 2007;81:4012–20. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–7. DOIPubMedGoogle Scholar
- Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev Med Virol. 2007;17:67–91. DOIPubMedGoogle Scholar
- Muller MA, Paweska JT, Leman PA, Drosten C, Grywna K, Kemp A, Coronavirus antibodies in African bat species. Emerg Infect Dis. 2007;13:1367–70.PubMedGoogle Scholar
- Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14:626–31. DOIPubMedGoogle Scholar
- Dominguez SR, O’Shea TJ, Oko LM, Holmes KV. Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis. 2007;13:1295–300.PubMedGoogle Scholar
- Proches S. The world’s biogeographical regions: cluster analyses based on bat distributions. J Biogeogr. 2005;32:607–14. DOIGoogle Scholar
- Mayer F, Dietz C, Kiefer A. Molecular species identification boosts bat diversity. Front Zool. 2007;4:4. DOIPubMedGoogle Scholar
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–45. DOIPubMedGoogle Scholar
- Swensson J. Bushmeat trade in Techiman, Ghana, West Africa. Uppsala (Sweden): Uppsala University; 2005.
- Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis. 2009;15:482–5. DOIPubMedGoogle Scholar
- de Souza Luna LK, Heiser V, Regamey N, Panning M, Drexler JF, Mulangu S, Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J Clin Microbiol. 2007;45:1049–52. DOIPubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. DOIPubMedGoogle Scholar
- Gelman A, Rubin DB. Markov chain Monte Carlo methods in biostatistics. Stat Methods Med Res. 1996;5:339–55. DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- Vallo P, Guillén-Servent A, Benda P, Pires DB, Koubek P. Variation of mitochondrial DNA reveals high cryptic diversity in Hipposideros caffer complex. Acta Chiropt. 2008;10:193–206. DOIGoogle Scholar
- Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol. 2007;81:1574–85. DOIPubMedGoogle Scholar
- Vijgen L, Keyaerts E, Moes E, Thoelen I, Wollants E, Lemey P, Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol. 2005;79:1595–604. DOIPubMedGoogle Scholar
- Pyrc K, Dijkman R, Deng L, Jebbink MF, Ross HA, Berkhout B, Mosaic structure of human coronavirus NL63, one thousand years of evolution. J Mol Biol. 2006;364:964–73. DOIPubMedGoogle Scholar
- Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol. 2006;23:7–9. DOIPubMedGoogle Scholar
- Carrington CV, Foster JE, Zhu HC, Zhang JX, Smith GJ, Thompson N, Detection and phylogenetic analysis of group 1 coronaviruses in South American bats. Emerg Infect Dis. 2008;14:1890–3. DOIPubMedGoogle Scholar
- Dobson AP. Virology. What links bats to emerging infectious diseases? Science. 2005;310:628–9. DOIPubMedGoogle Scholar
- Baumgarte S, de Souza Luna LK, Grywna K, Panning M, Drexler JF, Karsten C, Prevalence, types, and RNA concentrations of human parechoviruses, including a sixth parechovirus type, in stool samples from patients with acute enteritis. J Clin Microbiol. 2008;46:242–8. DOIPubMedGoogle Scholar
- Drexler JF, Luna LK, Stocker A, Almeida PS, Ribeiro TC, Petersen N, Circulation of 3 lineages of a novel Saffold cardiovirus in humans. Emerg Infect Dis. 2008;14:1398–405. DOIPubMedGoogle Scholar
- Takanashi S, Hashira S, Matsunaga T, Yoshida A, Shiota T, Tung PG, Detection, genetic characterization, and quantification of norovirus RNA from sera of children with gastroenteritis. J Clin Virol. 2009;44:161–3. DOIPubMedGoogle Scholar
- Forster JL, Harkin VB, Graham DA, McCullough SJ. The effect of sample type, temperature, and RNAlater on the stability of avian influenza virus RNA. J Virol Methods. 2008;149:190–4.PubMedGoogle Scholar
- Uhlenhaut C, Kracht M. Viral infectivity is maintained by an RNA protection buffer. J Virol Methods. 2005;128:189–91. DOIPubMedGoogle Scholar
- Becker MM, Graham RL, Donaldson EF, Rockx B, Sims AC, Sheahan T, Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc Natl Acad Sci U S A. 2008;105:19944–9. DOIPubMedGoogle Scholar
- Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. DOIPubMedGoogle Scholar
- Bredenbeek PJ, Snijder EJ, Noten FH, den Boon JA, Schaaper WM, Horzinek MC, The polymerase gene of corona- and toroviruses: evidence for an evolutionary relationship. Adv Exp Med Biol. 1990;276:307–16.PubMedGoogle Scholar
- Salemi M, Fitch WM, Ciccozzi M, Ruiz-Alvarez MJ, Rezza G, Lewis MJ. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J Virol. 2004;78:1602–3. DOIPubMedGoogle Scholar
- Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. DOIPubMedGoogle Scholar
- Sanchez CM, Gebauer F, Sune C, Mendez A, Dopazo J, Enjuanes L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology. 1992;190:92–105. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 15, Number 9—September 2009
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Christian Drosten, Institute of Virology, University of Bonn Medical Centre, Sigmund Freud-Strasse 25, 53127 Bonn, Germany;
Top