Volume 20, Number 7—July 2014
Dispatch
Detection and Genetic Characterization of Deltacoronavirus in Pigs, Ohio, USA, 2014
Cite This Article
Citation for Media
Abstract
In Ohio, United States, in early 2014, a deltacoronavirus was detected in feces and intestine samples from pigs with diarrheal disease. The complete genome sequence and phylogenetic analysis of the virus confirmed that the virus is closely related to a porcine deltacoronavirus (porcine coronavirus HKU15) reported in Hong Kong in 2012.
Coronaviruses (order Nidovirales, family Coronaviridae, subfamily Coronavirinae) are single-stranded, positive-sense, enveloped RNA viruses with a genome size ranging from 25.4 to 31.7 kb (1). Coronaviruses traditionally were classified into groups 1, 2, and 3 on the basis of their antigenic relationships (2). The traditional classification recently was replaced by 4 genera (Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus), as described by the International Committee for Taxonomy of Viruses (http://www.ictvonline.org/virusTaxonomy.asp?msl_id = 27). Virus from each coronavirus genus has been found in diverse host species, including mammals and birds (3). Viruses of the Alphacoronavirus, Betacoronavirus, and Deltacoronavirus genera have been detected in swine (3). Transmissible gastroenteritis virus and porcine epidemic diarrhea virus (PEDV) are members of the genus Alphacoronavirus; these viruses cause severe diarrhea in swine herds, leading to significant economic loss in many countries, including the United State (4–6). There is little information about deltacoronavirus infections in pigs, and only 1 surveillance study from Hong Kong reported its detection in pigs (1). The virus has not been reported to be associated with clinical disease in these animals. We report the detection and genetic characterization of a deltacoronavirus in pigs from farms in Ohio, United States; the pigs all had clinical diarrheal disease.
During the end of January and the beginning of February in 2014, feces and intestine samples from pigs on 5 Ohio farms were submitted to the Animal Disease Diagnostic Laboratory at the Ohio Department of Agriculture. The farm managers reported outbreaks of diarrheal disease in sows and piglets. The clinical signs were similar to those associated with PEDV infection, including watery diarrhea in sows and death in piglets. However, the death rate in piglets (30%–40%) was lower than that typically observed with PEDV infection. Test results for samples from Farm 1 were negative for PEDV, transmissible gastroenteritis virus, rotavirus, and Salmonella spp. Examination of the samples by electron microscopy showed that most contained coronavirus-like virus particles. This finding prompted the laboratory to look further for the presence of viruses other than alphacoronaviruses.
Because deltacoronavirus has been reported in pigs (1), we designed a deltacoronavirus-specific reverse transcription PCR (RT-PCR). RNA was extracted from feces and intestine samples by using the TRIzol (Invitrogen, Carlsbad, CA, USA) method, and RT-PCR was performed by using the QIAGEN OneStep RT-PCR kit (QIAGEN, Hilden, Germany) with porcine deltacoronavirus–specific primers for membrane (M) gene 67F(5′-ATCCTCCAAGGAGGCTATGC-3′), 560R(5′-GCGAATTCTGGATCGTTGTT-3′), and nucleocapsid (N) gene 41F(5′-TTTCAGGTGCTCAAAGCTCA-3′), 735R(5′-GCGAAAAGCATTTCCTGAAC-3′). These primers were designed by using the conserved regions of 2 available porcine deltacoronavirus sequences (1). RT-PCR was performed under the following cycling conditions: 50°C for 30 min and 95°C for 15 min for the RT reaction, followed by 40 cycles of amplification at 95°C for 15 s, 55°C for 45 s, and 72°C for 1 min, with a final extension at 72°C for 7 min. All samples (3 piglet small intestines and 9 feces samples) from Farm 1 were positive for deltacoronavirus by the 2 RT-PCR assays for M and N genes (Table 1). Nucleotide sequences were determined for both amplified M and N fragments. A BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) search of the sequences of both M and N fragments showed 99% nt identity with the porcine coronavirus HKU15 (PorCoV HKU15)-155. Therefore, the virus detected in this study was PorCoV HKU15, which belongs to the Deltacoronavirus genus.
RT-PCR was then run on samples from Farms 2–5. The results from the 5 farms are summarized in Table 1. Of the total 42 samples from the 5 farms, 39 (92.9%) were positive for PorCoV HKU15 by RT-PCR (Table 1). In addition, 5 (11.9%) of the 42 samples were positive for classical US PEDV instead of a recently reported variant PEDV (7) (Table 1). None of the samples tested were positive for transmissible gastroenteritis virus. Four (9.5%) of the 42 samples were positive for PorCoV HKU15 and PEDV, indicating that mixed infection with PorCoV HKU15 and PEDV occurred in some pigs.
On the basis of 2 complete genome sequences from GenBank, of PorCoV HKU15-44 and HKU15-155, we designed 16 pairs of primers to determine the whole genome of extracted RNA samples (OH1987) from Farm 1 (Table 2). The genome of PorCoV HKU15 OH1987 comprised 25,422 nt (GenBank accession no. KJ462462). The genome organization and the transcription regulatory sequence motif 5′-ACACCA-3′ of PorCoV HKU15 OH1987 are the same as those reported for PorCoV HKU15-155 (1). Similar to the BLAST search results of partial N and M fragments, the BLAST search of the whole genome of the PorCoV HKU15 OH1987 showed 99% nt identity to PorCoV HKU15-155. BLAST search of the spike gene of PorCoV HKU15 OH1987 showed 99% nt identity to PorCoV HKU15-44. These results confirmed that the coronavirus detected was a deltacoronavirus.
Figure 1
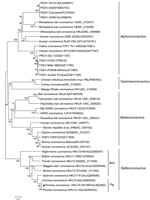
Figure 1. Phylogenetic tree constructed on the basis of the whole-genome sequences of virus strains from 4 coronavirus genera (Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus), including the porcine coronavirus HKU15 OH1987 strain (indicated with...
Figure 2

Figure 2. Phylogenetic analyses of spike protein (A) and nucleocapsid protein (B) of virus strains of 4 coronavirus genera (Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus), including the porcine coronavirus HKU15 OH1987 strain (indicated with...
Phylogenetic analysis of the complete genome of PorCoV HKU15 OH1987 showed that the OH1987 strain clustered with the other 2 PorCoVs, HKU15-155 and HKU15-44, and was distinct from the bird deltacoronaviruses (Figure 1). In addition, the phylogenetic trees constructed by using the amino acid sequences of the spike glycoprotein and nucleocapsid protein showed that the OH1987 virus clustered with HKU15-155 and HKU15-44 (Figure 2); this finding is in agreement with that in a previous study (1). The OH1987 virus differs from HKU15-155 in the spike gene at nt 19469 and in the noncoding region at nt 25044; a 3-nt insertion is present at each location, making the whole-genome sequence of the OH1987 virus 6 nt longer than that of HKU15-155. The 2 insertion sites of the virus are identical to those of the HKU15-44 strain of porcine deltacoronavirus. However, the genome of OH1987 virus is 1 nt longer than that of HKU15-44 because of the insertion site at nt 25263, located in 3′ untranslated region. Of interest, strain OH1987 was most closely related to HKU15-155 when whole-genome sequence was used for phylogenetic analysis (Figure 1), but strain OH1987 was more closely related to HKU15-44 when phylogenetic analysis of spike protein and nucleocapsid protein was performed (Figure 2). On basis of the partial genome sequence, strain OH1987 was also closely related to a deltacoronavirus found in the Asian leopard cat (the whole-genome sequence is not available in GenBank).
We detected a porcine deltacoronavirus in pigs in the United States. Although the genetic and phylogenetic analyses showed that the newly emergent strain, PorCoV HKU15 OH1987, was closely related to 2 strains from China, HKU15-155 and HKU15-44, in the genus Deltacoronavirus, when and how this virus was introduced into United States remain unknown. Further investigation is needed to determine whether infection with PorCoV HKU15 results in disease in pigs and if the virus was responsible for the clinical disease observed in outbreaks on the 5 Ohio farms in this study. In addition, surveillance should be conducted in other US states to define the distribution of the virus among the US pig population. Moreover, whole-genome sequence analysis should be performed for other strains from different locations to determine whether the virus was introduced into the United States by a single entry or by multiple entries.
Dr Wang is a laboratory scientist at the Animal Disease Diagnostic Laboratory, Ohio Department of Agriculture. His research interests include infectious diseases, molecular virology, and vaccine development.
Acknowledgment
We acknowledge and appreciate the excellent technical help provided by Jason Herr and Kerri Lawrence.
References
- Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86:3995–4008 . DOIPubMedGoogle Scholar
- Woo PC, Huang Y, Lau SK, Yuen KY. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2:1804–20 . DOIPubMedGoogle Scholar
- Chan JF, To KK, Tse H, Jin DY, Yuen KY. Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 2013;21:544–55. DOIPubMedGoogle Scholar
- Schwegmann-Wessels C, Herrler G. Transmissible gastroenteritis virus infection: a vanishing specter. Dtsch Tierarztl Wochenschr. 2006;113:157–9 .PubMedGoogle Scholar
- Song D, Park B. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes. 2012;44:167–75. DOIPubMedGoogle Scholar
- Huang YW, Dickerman AW, Pineyro P, Li L, Fang L, Kiehne R, Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. MBio. 2013;4:e00737–13. DOIPubMedGoogle Scholar
- Wang L, Byrum B, Zhang Y. New variant of porcine epidemic diarrhea virus, United States [letter]. 2014. Emerg Infect Dis. 2014;20:917–9 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors were co–principal investigators.
Table of Contents – Volume 20, Number 7—July 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Yan Zhang, Animal Disease Diagnostic Laboratory, Ohio Department of Agriculture, 8995 East Main St, Bldg. 6, Reynoldsburg, OH 43068, USA
Top