Volume 21, Number 4—April 2015
Dispatch
Candidate New Rotavirus Species in Sheltered Dogs, Hungary
Cite This Article
Citation for Media
Abstract
We identified unusual rotavirus strains in fecal specimens from sheltered dogs in Hungary by viral metagenomics. The novel rotavirus species displayed limited genome sequence homology to representatives of the 8 rotavirus species, A–H, and qualifies as a candidate new rotavirus species that we tentatively named Rotavirus I.
Rotaviruses (family Reoviridae, genus Rotavirus) are major causes of acute dehydrating gastroenteritis in birds and mammals (1). Rotaviruses have an 11-segmented dsRNA genome encoding 6 structural proteins (viral protein [VP] 1–4, VP6, and VP7) and at least 5 functional nonstructural proteins (NSPs; NSP1–NSP5) (Technical Appendix Table 1). Traditionally, rotaviruses have been classified into (sero)groups on the basis of major antigenic differences that predominantly reside in the VP6 and of the genomic RNA profile obtained by polyacrylamide gel electrophoresis and silver staining (1). Recently, a VP6 gene sequence–based classification scheme has been proposed to replace the conventional methods. An empirical 53% aa identity was demonstrated to reliably distinguish strains of various rotaviruses groups (2). Also, reclassification of the 8 rotavirus groups as distinct species within the Rotavirus genus, designated Rotavirus A–H, has been proposed.
Rotavirus A has been detected in a wide variety of mammals and birds. In mammals, both endemic and epidemic forms of rotavirus B, C, E, and H infections have been described, whereas rotavirus D, F, and G have been identified only in birds (1–3). Genetically diverse rotaviruses have been found in some viral metagenomics studies (4,5). Using the metagenomic approach and the VP6-based molecular classification scheme, we found evidence for a novel rotavirus species that we tentatively called Rotavirus I.
During 2012, we collected fecal specimens from sheltered dogs in northern Hungary to detect enteric viruses. Of 63 samples obtained from 50 animals, 37 randomly selected samples (from 33 animals) were subjected to random primed reverse transcription PCR and semiconductor sequencing by using the Ion Torrent PGM platform (New England Biolabs, Ipswich, MA, USA) (Technical Appendix). Bioinformatics analysis consisted of the mapping of reads >40 bases against ≈1.7 million viral sequences downloaded from GenBank by applying moderately rigorous mapping parameters (length fraction 0.6; similarity fraction 0.8) within the CLC Genomics Workbench (http://www.clcbio.com/).
One sample (KE135/2012) obtained from a suckling dog in May 2012 was positive for several enteric viruses. When analyzing the initially obtained ≈60.5-K sequence reads, in addition to canine rotavirus A (141 reads), astrovirus (2,399 reads), and parvovirus (3,623 reads), we identified a single 53-nt sequence read that mapped to the VP1 gene of rotavirus B. Another sample, KE528/2012, collected during August 2012 from an adult dog with diarrhea, was positive for coronavirus (30 reads), vesivirus (17 reads), picodicistrovirus (3 reads), and astrovirus (1 read); in addition, 7 and 5 sequence reads, respectively, mapped to the VP1 and VP3 genes of rotavirus H and/or B.
Subsequently, we enriched genomic dsRNA of KE135/2012 by differential LiCl precipitation; however, the enriched dsRNA remained invisible by polyacrylamide gel electrophoresis and silver staining. Because of the apparent low titer of the novel rotavirus, we tried to obtain more sequence data by drastically increasing the output in parallel sequencing runs. De novo assembly of the resulting ≈1.59 million sequence reads readily identified homologs of the structural and some nonstructural genes, which were divergent from rotavirus A–H reference sequences (Table; Technical Appendix Table 1). Determination of the coding regions in most cases was successful by extension of the termini of consensus sequences using the Ion Torrent sequence reads. However, we found no evidence for NSP3 and NSP4 with this approach, probably because of the great sequence divergence of these genes across members of the genus (6,7). Because the genomic RNA of each rotavirus species is characterized by low GC (guanine:cytosine) content (29%–40%), we expected that contigs with low GC content and with no GenBank homologs might be good candidates for detecting the missing genes. Indeed, further assembly and subsequent analysis of selected sequence stretches helped to identify the NSP3 by similarity search through the blastx engine (http://blast.ncbi.nlm.nih.gov/Blast.cgi) after an 800-bp long fragment was obtained, and analysis of the structural features of the deduced protein sequence supported detection of the putative NSP4. The obtained consensus sequence was used as reference to map other viral metagenomics data generated from the sheltered dog population; however, except for the aforementioned sample, KE528/2012, we found no additional specimens by this method to contain homologous viruses. The 2 related unusual rotaviruses, KE135/2012 and KE528/2012, had conserved genome segment termini (5′ end, GGC/TA; 3′ end, AACCC) and shared high sequence identities in most genes (e.g., VP2: 88% nt, 95% aa; NSP4: 99% nt, 99% aa) and very low sequence similarity in the VP7 gene (53% nt, 38% aa) (GenBank accession nos. KM369887–KM369908; Technical Appendix).
The deduced VP6 amino acid sequences served as the basis to classify these 2 unusual rotavirus strains (2). The greatest amino acid sequence identity of the VP6 proteins was found when compared to the novel rotavirus H strains (<46%); lower sequence similarities were found in comparison to randomly selected representatives of other rotvirus species (e.g., rotavirus G and B, <37%; rotavirus A, C, D, and F, <18%).
Figure 1

Figure 1. Structure comparison of rotavirus viral protein (VP) 6 proteins. A) Structure-based amino acid sequence alignment of the novel canine rotavirus VP6 protein and the template bovine rotavirus A VP6 protein. The...
Figure 2
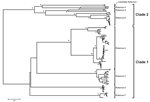
Figure 2. Protein sequence–based phylogenetic tree of the rotavirus viral protein 6 gene obtained by the neighbor-joining algorithm. Asterisks indicate >90% bootstrap values. The 2 canine rotavirus strains from Hungary that belong to...
To extend the analysis and assess whether the obtained VP6 gene might be functionally integral, we conducted molecular modeling of the amino acid sequence. In brief, amino acid sequence similarity values created a reliable protein model (8,9) showing similar protein folding of the VP6 monomer and comparable electrostatic charge pattern around the 3-fold axis of the VP6 homotrimer to that experimentally determined for rotavirus A (Figure 1). Subsequent phylogenetic analysis of the VP6 protein identified 2 major clades of rotaviruses (6). The novel rotavirus strains clustered with species H, G, and B within clade 2, whereas clade 1 included representative strains of species A, C, D, and F (Figure 2). This pattern of clustering was also evident when we analyzed the remaining genes. Collectively, sequence and phylogenetic analysis demonstrated moderate genetic relatedness of the unusual canine rotaviruses to representative strains of species A–H, suggesting that they belong to a novel species, tentatively called Rotavirus I. The prototype strains were named RVI/Dog-wt/HUN/KE135/2012/G1P1 and RVI/Dog-wt/HUN/KE528/2012/G2P1 according to recent guidelines (10) (online Technical Appendix).
Short rotavirus sequences detected recently in the fecal viral flora of cats and California sea lions (4,5) showed closer relatedness to our strains in the amplified VP6- and VP2-specific stretches, respectively, than to the corresponding genomic regions of reference rotavirus species (VP6, ≈70 aa, 67% vs. <55%; VP2, ≈160 aa, 78%–86% vs. <44%) (Technical Appendix). These published data (4,5) together with our results suggest that genetically related non–rotavirus A–H strains occur in various carnivore host species and geographic settings.
We identified 2 representative strains of a novel rotavirus species, Rotavirus I. Many questions remain, including those related to the epidemiology, host range, and evolution of this species. One intriguing finding was the distantly related VP7 genes expressed on a fairly conserved genetic backbone. Typically, very low sequence identity values within the VP7 gene (e.g., rotavirus A, as low as 60% nt and 55% aa; rotavirus B, 54% nt, 46% aa; rotavirus H, 63% nt, 56% aa) can be seen when strains from different host species are compared (11–13). Whether the VP7 gene(s) of rotavirus I strains could have been acquired in the past from another rotavirus species by reassortment remains uncertain, given that reassortment among various rotavirus species is thought to occur only rarely (7,14). Further information is needed to better understand this genetic diversity within rotavirus I.
Dr. Mihalov-Kovács is a PhD student at the Pathogen Discovery Group, Institute for Veterinary Medical Research, Centre for Agricultural Research, Hungarian Academy of Sciences. Her research interests include discovery of novel viruses in domesticated animals.
Acknowledgment
Financial support was obtained from the Momentum Program (Hungarian Academy of Sciences) and the Hungarian Scientific Research Program (OTKA [Országos Tudományos Kutatási Alapprogramok] 108793; licensing of the Schrödinger Suite software package). Á.G. received a János Bolyai fellowship; F.J. received additional funding from TÁMOP (4.2.4.A/2-11-1-2012-0001).
References
- Estes MK, Kapikian AZ. Rotaviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, et al., editors. Fields virology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 1917–74.
- Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Van Ranst M, Johne R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch Virol. 2012;157:1177–82. DOIGoogle Scholar
- Marthaler D, Rossow K, Culhane M, Goyal S, Collins J, Matthijnssens J, Widespread rotavirus H in commercially raised pigs, United States. Emerg Infect Dis. 2014;20:1195–8 . DOIGoogle Scholar
- Ng TF, Mesquita JR, Nascimento MS, Kondov NO, Wong W, Reuter G, Feline fecal virome reveals novel and prevalent enteric viruses. Vet Microbiol. 2014;171:102–11. DOIGoogle Scholar
- Li L, Shan T, Wang C, Côté C, Kolman J, Onions D, The fecal viral flora of California sea lions. J Virol. 2011;85:9909–17. DOIGoogle Scholar
- Kindler E, Trojnar E, Heckel G, Otto PH, Johne R. Analysis of rotavirus species diversity and evolution including the newly determined full-length genome sequences of rotavirus F and G. Infect Genet Evol. 2013;14:58–67. DOIGoogle Scholar
- Trojnar E, Otto P, Roth B, Reetz J, Johne R. The genome segments of a group D rotavirus possess group A–like conserved termini but encode group-specific proteins. J Virol. 2010;84:10254–65. DOIGoogle Scholar
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. DOIGoogle Scholar
- Mathieu M, Petitpas I, Navaza J, Lepault J, Kohli E, Pothier P, Atomic structure of the major capsid protein of rotavirus: implications for the architecture of the virion. EMBO J. 2001;20:1485–97. DOIGoogle Scholar
- Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Bányai K, Brister JR, Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch Virol. 2011;156:1397–413. DOIGoogle Scholar
- Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1–like and bovine rotavirus strains. J Virol. 2008;82:3204–19 and. DOIGoogle Scholar
- Marthaler D, Rossow K, Gramer M, Collins J, Goyal S, Tsunemitsu H, Detection of substantial porcine group B rotavirus genetic diversity in the United States, resulting in a modified classification proposal for G genotypes. Virology. 2012;433:85–96. DOIGoogle Scholar
- Wakuda M, Ide T, Sasaki J, Komoto S, Ishii J, Sanekata T, Porcine rotavirus closely related to novel group of human rotaviruses. Emerg Infect Dis. 2011;17:1491–3 .
- Esona MD, Mijatovic-Rustempasic S, Conrardy C, Tong S, Kuzmin IV, Agwanda B, Reassortant group A rotavirus from straw-colored fruit bat (Eidolon helvum). Emerg Infect Dis. 2010;16:1844–52.
Figures
Table
Cite This ArticleTable of Contents – Volume 21, Number 4—April 2015
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Krisztián Bányai, Institute for Veterinary Medical Research, Centre for Agricultural Research, Hungarian Academy of Sciences, H-1143 Budapest, Hungária krt. 21, Hungary
Top