Volume 22, Number 12—December 2016
Synopsis
Assessing the Epidemic Potential of RNA and DNA Viruses
Cite This Article
Citation for Media
Abstract
Many new and emerging RNA and DNA viruses are zoonotic or have zoonotic origins in an animal reservoir that is usually mammalian and sometimes avian. Not all zoonotic viruses are transmissible (directly or by an arthropod vector) between human hosts. Virus genome sequence data provide the best evidence of transmission. Of human transmissible virus, 37 species have so far been restricted to self-limiting outbreaks. These viruses are priorities for surveillance because relatively minor changes in their epidemiologies can potentially lead to major changes in the threat they pose to public health. On the basis of comparisons across all recognized human viruses, we consider the characteristics of these priority viruses and assess the likelihood that they will further emerge in human populations. We also assess the likelihood that a virus that can infect humans but is not capable of transmission (directly or by a vector) between human hosts can acquire that capability.
A series of recent emerging infectious disease outbreaks, including the 2014 Ebola virus disease (EVD) epidemic in West Africa and the continuing Zika virus disease epidemic in the Americas, have underlined the need for better understanding of which kinds of pathogens are most likely to emerge and cause disease in human populations. Many, although not all, emerging infectious diseases are caused by viruses, and these frequently emerge from nonhuman host reservoirs (1–3). The enormous diversity (4) and high rates of evolution (5) of viral pathogens discourage attempts to predict with any precision which ones are most likely to emerge in humans. However, there is some consensus, at least in general terms, regarding the kinds of traits that are most essential in determining the capacity of a virus to infect, cause disease, and spread within human populations (Table 1). We focus on one of these traits, the capacity of a virus to spread from one human to another (by any transmission route other than deliberate laboratory exposure), a key determinant of the epidemic potential of a virus.
A theoretical framework for studying the dynamics of infectious disease outbreaks is well established (6). The capacity of an infectious disease to spread in a host population can be quantified in terms of its basic reproduction number, R0. R0 is defined as the average number of secondary cases generated by a single primary case in a large, previously unexposed host population, and its value tells us a great deal about the epidemiology of a pathogen. R0 = 0 indicates no spread in that population; this value would apply to zoonotic infections that do not spread between humans. R0 in the range 0<R0<1 indicates that chains of transmission are possible but that outbreaks will ultimately be self-limiting. R0>1 indicates that major epidemics can occur or that the disease may become endemic in that host population. A higher value of R0 also indicates that a greater reduction in transmission rates must be achieved to control an epidemic (6). R0 values have been estimated for >60 common human pathogens (7), including human influenza A virus (R0<2), measles virus (R0<18), and dengue virus (R0<22).
R0 is determined by a combination of pathogen traits, such as its transmission biology, which is itself a complex interplay between the within-host dynamics of the pathogen and the host response to infection, and host traits, such as demography, behavior, genetics, and adaptive immunity. Consequently, for any given infectious disease, R0 can vary between host species and between host populations. Infectious diseases with R0 close to 1 are a particular concern because small changes in their epidemiologies can lead to major changes in the threat they pose to public health (8).
Figure 1
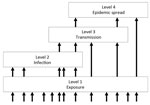
Figure 1. Pathogen pyramid for RNA and DNA viruses. Level 1 indicates viruses to which humans are exposed but which do not infect humans. Level 2 indicates viruses that can infect humans but...
R0 is closely related to another conceptual approach to disease emergence, the pathogen pyramid. There are different versions of this scheme (3,9). We consider a pyramid of 4 levels (Figure 1). Level 1 represents the background chatter of pathogens to which humans are continually or sporadically exposed but most of which are not capable of causing infection. Other levels can be considered in terms of the R0 of the pathogen in humans: level 2 corresponds to R0 = 0, level 3 to 0<R0<1, and level 4 to R0>1.
Identifying and Characterizing Level 3 and 4 Viruses
We updated our previous systematic literature review (10) of the capacity of virus species to transmit between humans (i.e., level 3 and level 4 viruses; Technical Appendix). Such viruses are found in 25 of 29 families containing viruses that infect mammals or birds (discounting 2 reports of family Nodaviridae species in mammals/birds). The 4 exceptions comprise 2 families that have no known human-infective viruses (Arteriviridae and Birnaviridae) and 2 with species that have been reported in humans but only at level 2 (Asfarviridae and Bornaviridae).
A total of 22 of these families contain level 4 viruses with epidemic potential in humans (sometimes described as human-adapted viruses) (11). This finding indicates that this capability is widely distributed among virus taxa. The 3 families with level 3 viruses but no level 4 viruses are the Arenaviridae, Bunyaviridae, and Rhabdoviridae.
A list of 37 presumptive level 3 virus species is provided in Table 2. These species cover a wide taxonomic range and a variety of transmission routes, including vectorborne. Several level 3 viruses have historically been associated with sizeable outbreaks (>100 cases) in human populations: Bwamba, Oropouche, Lake Victoria Marburg, Sudan Ebola, and o’nyong-nyong viruses. For some other viruses, including Guanarito, Junin, lymphocytic choriomeningitis, and Sabia (all arenaviruses); simian virus 40; Titi monkey virus; and influenza A(H5N1) virus, human-to-human transmission is rare or merely suspected. In addition, several viruses are only known or believed to transmit between humans by iatrogenic routes or vertical transmission; this group (Table 2) might be regarded as unlikely epidemic threats.
When a virus is transmitted by a vector, it can be particularly difficult to confirm or exclude the infectiousness of human cases, as with Semliki forest, Barmah forest, and Rift Valley fever viruses. Similarly, even when human–vector–human transmission is believed to occur, it is often difficult to quantify its contribution to a given outbreak, as with Venezuelan equine encephalitis virus.
Level 2 viruses are those that can infect humans (>100 species) but have never been reported to be transmitted by humans (10). In at least some instances, such as influenza A(H5N1) virus (12), this finding is attributable to tissue tropisms during human infection that are incompatible with onward transmission.
Shifts in pyramid level equate to shifts in the public health threat posed by a virus. We consider possible shifts in the following sections.
Level 1 to Levels 3 and 4
Virus species of mammalian and, more rarely, avian origin are sometimes observed to be transmissible between humans when first found in humans, which constitutes a jump from level 1 straight to level 3 or 4 (Figure 1), and events of this kind have been reported regularly. Recent examples that appear on the basis of available evidence to fit this model include severe acute respiratory syndrome coronavirus (first reported in humans in 2003), Bundibugyo Ebolavirus (2008), Lujo virus (2009), severe fever with thrombocytopenia syndrome virus (2011), and Middle East respiratory syndrome coronavirus (MERS-CoV) (2012).
We still have incomplete knowledge of the diversity of viruses that infect mammals and birds; the few hundred recognized species (4) surely represent only a small fraction of the total (3). Moreover, we have few predictors of potential human-to-human transmissibility. One possible indicator is emergence from nonhuman primates, with suggestions that primate viruses are more likely to be able to, or to acquire the ability to, spread in human populations (13,14). However, emergence of human transmissible viruses from bat (e.g., severe acute respiratory syndrome coronavirus) or bird (e.g., influenza) reservoirs indicates that this trait is associated with a wide range of reservoirs.
Level 2 to Levels 3 and 4
The possibility that level 2 viruses might acquire the capacity to be transmitted between humans (i.e., move into level 3 or 4) is a major concern, especially in the context of influenza A(H5N1) virus and other avian influenza virus subtypes. However, there are few examples of this transition throughout the entire recorded history of human viruses going back to 1901. One possible example involves the simian immunodeficiency virus (SIV) and HIV. A SIVsmm-derived laboratory strain of SIV has been reported to infect humans, but without onward transmission (15). SIVsmm is related to HIV-2. SIVcpz, which is related to HIV-1, has not been directly observed in humans. However, different HIV-1 lineages, independently derived from SIVcpz, are variably transmissible in humans, and the pandemic HIV-1 M lineage was the only virus to overcome a key host restriction factor (human tetherin) (16). The only other examples of viruses newly transmitted between humans relate to rare instances of iatrogenic transmission (e.g., Colorado tick fever or rabies viruses).
Epidemiologic and phylogenetic considerations routinely inform our assessment of the likelihood of human-to-human transmission being observed in the future. For example, there is markedly less concern about rabies virus than about avian influenza virus, and we suggest 2 reasons for this observation. First, rabies virus has a much longer history of and a much higher incidence of human infection, but human-to-human transmission is extremely rare. Second, there is no evidence that other rhabdoviruses viruses (with the possible exception of Bas-Congo virus, which represents a novel genus) are transmissible in humans (or primates more generally).
Level 3 to Level 4
Level 3 viruses can also become level 4 viruses. We note that virus evolution is not (necessarily) required for R0 to become >1 in human populations. Differences in host (or vector) behavior, ecology, or demography might be sufficient (8).
Instances of shifts from level 3 to level 4 in recent times have been infrequent. Three candidates are Ebolavirus, Zika virus, and chikungunya virus. However, although these viruses have caused epidemics of unprecedented size in humans populations in the past decade, the condition R0>1 in human populations might had been previously met for all 3 viruses (17–19).
For Ebola virus, the epidemic in West Africa in 2014 constituted the first appearance of this virus in high-density, urban populations, which is expected to correspond to a higher value of R0. The chikungunya virus epidemic in the Indian Ocean region in 2005 was associated with a vector species jump (from Aedes aegypti to Ae. albopictus mosquitoes) that has been linked to a mutation in the virus envelope 1 protein gene (18). The chikungunya virus epidemic in the Caribbean region in 2013 followed the first appearance of chikungunya in the Americas and infected populations that had no history of exposure to the virus. The current Zika virus epidemic in South America appears to be another example of a transition from a level 3 to a level 4 arbovirus associated with geographic spread into areas with high densities of vectors (19). Occasional Zika virus transmission directly from infected humans to other humans are of considerable interest, but probably contribute little to R0.
Chikungunya, Zika and the other level 4 arboviruses (yellow fever and dengue viruses) illustrate that, for arboviruses, a high potential for spread in human populations is linked to carriage by anthropophilic vector species, particularly mosquitoes of the genus Aedes. In contrast, no tick species are regarded as anthropophilic, and there are no level 4 and few level 3 tickborne arboviruses.
Epidemiologic Patterns
The preceding sections illustrate that identifying transitions of viruses between level 2 and level 3 or between level 3 and level 4 is not always straightforward. Standard epidemiologic theory can help clarify our expectations.
Figure 2
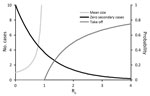
Figure 2. Expected outbreak dynamics for RNA and DNA viruses given a single primary case in a large, previously unexposed host population, as a function of the basic reproduction number R0. Mean size...
As we discussed, pyramid level is related to the basic reproduction number R0 in human populations. In turn, the value of R0 is indicative of expected outbreak dynamics. Some key results (Figure 2) are the probability that a single primary case will generate >1 secondary cases (for any value of R0), the expected average size of an outbreak generated (over the range 0<R0<1), and the probability that an epidemic will spread in the human population (for R0>1). These results strictly apply to homogeneous infections in a homogeneous host population, although more general frameworks can accommodate host or pathogen heterogeneity (20–22). Nonetheless, the key predictions that secondary cases do not always occur even if R0>0 and that major epidemics do not always occur even if R0>1 are robust.
Figure 3

Figure 3. Distribution of outbreak sizes for RNA and DNA viruses as plots of outbreak size x (horizontal axis) versus fraction of outbreaks of size >x (vertical axis), both on logarithmic scales....
From an epidemiologic perspective, our confidence that a putatively level 2 virus is truly incapable of human-to-human transmission is thus a function of the number of index cases observed. The transition between level 3 and level 4 can be studied in terms of the expected distribution of outbreak sizes (23). In the range 0<R0<1, an overdispersed distribution of outbreak sizes is expected: most outbreaks are small (often just single cases) with a long tail of larger outbreaks. This pattern has been reported for a range of emerging viral diseases (Figure 3). As the critical threshold R0 = 1 is approached, this value is signaled in the outbreak size distribution (Figure 3). This framework has been used successfully to monitor the epidemiology of measles virus in the United Kingdom after a decrease in childhood vaccination rates in the late 1990s and indicated the approach to the critical threshold that corresponded to loss of herd immunity (23).
Outbreak size distribution analysis has been applied to human case data for Andes virus (24), monkeypox virus (20), and MERS-CoV (25) (Figure 3). For EVD up to 2013, data are clearly inconsistent with theoretical expectation for R0 <1 (Figure 3), which suggests that large numbers of small outbreaks have remained undetected or that R0 was already >1 in at least some settings. Either way, R0 ≈1 for EVD in humans implies that small differences in the biology or epidemiology of the virus would lead to large changes in scale of outbreaks (8), which could make events such as the EVD epidemic in 2014, if not predictable, then much less unexpected.
Evolution
Changes in pyramid level might be mediated by virus evolution or changes in virus ecology (28). A major issue is whether the capacity of a virus to spread in human populations arises as a result of adaptation (evolution of transmissibility that occurs during human infection) or preadaptation (genetic variation within nonhuman reservoirs that predisposes a virus not only to infect humans but also transmit between humans, noting that RNA viruses often show high levels of genetic variation such that they are sometimes described as quasi-species [29]). These alternatives have been characterized as tailor-made and off-the-shelf, respectively (28). The first alternative implies a progression from no or low transmissibility between humans to moderate or high transmissibility. The second alternative implies moderate or high transmissibility at first infection of humans.
We consider that our survey of documented changes of pyramid level is most consistent with the off-the-shelf model of virus emergence. In particular, we can find no convincing examples of level 2 viruses becoming level 3 or 4 viruses, which suggests that, if this happens at all, it typically happens sufficiently rapidly (i.e., requires a sufficiently small number of introductions) that we fail to observe the level 2 phase. In contrast, we regularly observe viruses at levels 3 or 4 the first time they are detected in human populations.
Nonetheless, the possibility of virus evolution of transmissibility in a new host has been demonstrated experimentally for influenza A(H5N1) virus in ferrets (30). A theoretical study (31) suggested that the fact that this virus subtype has been circulating widely in poultry populations, with frequent human exposure and sporadic human infection for almost 20 years, provides little or no reassurance about its future evolutionary trajectory.
HIV lineages show clear evidence of adaptation to humans (16), but as discussed earlier, it is not clear whether the SIV lineages that gave rise to HIV-1 or HIV-2 were capable of transmission between human hosts. We speculate that extended infection times make tailor-made emergence more likely for retroviruses.
Transmission
Demonstrating that an infected human has the potential to transmit the infection to another human is not always straightforward. High virus titers in body secretions and excretions, blood, or skin are considered indicative. Case clusters are suggestive, but if persons occupy the same environment (e.g., household), then it might be difficult to rule out common exposure. Case clusters must be epidemiologically plausible (i.e., delimited in space and time in a manner consistent with the known or assumed epidemiology of the virus). Genotyping techniques are useful tools for confirming a cluster but do not resolve the source of infection.
For several of the viruses we studied (e.g., Bas-Congo, Lujo, Nelson Bay, and severe fever with thrombocytopenia syndrome viruses) (Table 2), the evidence for human-to-human transmission is best regarded as tentative, particularly where putative clusters were small. Such assessments can be even more difficult for vectorborne viruses. In many situations, the best evidence for the human-to-human transmission will come from analysis of virus genome sequences.
Phylogenetic Analysis
One approach to resolving the question of human-to-human transmission is analysis of nucleotide sequence data, sometimes referred to as forensic phylogenetics. Nucleotide substitution rates in fast-evolving RNA viruses, such as MERS-CoV and Ebola virus, are ≈1–5 × 10−3/site/year (32,33), making it possible to use sequences isolated from different hosts at different times to estimate time-resolved phylogenetic trees. Estimates of the transmission chain from temporal sequence data can be improved by incorporating additional information on the date of onset of individual cases, duration of latent and infectious periods, and overall prevalence (34).
Figure 4

Figure 4. Phylogenetic trees for simulated emerging infectious disease outbreaks caused by RNA and DNA viruses in a mixed population of 1,000 human and 5,000 nonhuman hosts. Trees were constructed by using a...
We provide some example phylogenetic trees generated from simulated epidemics (Figure 4). In an epidemic in an animal reservoir with occasional transmission to humans (Figure 4, panel A), for each human sequence, the most closely related next sequence is of animal origin. Clusters of closely related human sequences are shown, and the distribution of the expected cluster sizes is a function of R0 (Figure 4, panels B, C) (35).
In an outbreak, it might be difficult to find and sample the putative source animal cases. However, estimating the time to most recent common ancestor (TMRCA) of the human cases will indicate how long the infection has been spreading. For sporadic zoonoses (Figure 4, panel A), most transmission has occurred unobserved in the animal reservoir, and the TMRCA of pairs of human cases will be long because these sequences are not closely related. For outbreaks involving human-to-human transmission (Figure 4, panels B, C), the TMRCA of the cluster of human cases will be closer to the date of the first human infection (whether sampled or not) and provides the estimated date of the zoonotic event.
Use of sequence data to distinguish between multiple instances of human infection from a common animal source and human-to-human transmission in the early stages of an outbreak is extremely challenging because of short timescales, and involvement of few mutations. However, genetic differences and phylogenetic evidence show that at least 2 of the first 3 reported cases of influenza A (H7N9) virus infection in humans were believed to originate from distinct domestic avian sources (36).
Further sequencing of avian samples implied that a low-pathogenicity influenza A(H7N9) virus strain had been spreading in domestic birds for ≈1 year before sporadic cases were detected in humans (37). Similarly, detection of genetically distant lineages of MERS-CoV, which persisted for only a few months each, suggest multiple introductions from an animal reservoir and only limited human-to-human transmission to date (32). In contrast, the influenza A(H1N1) pandemic in 2009 and the EVD epidemic in West Africa in 2014 were believed to be the results of single zoonotic events, followed by sustained human-to-human transmission (33), as shown by a single rapidly expanding lineage.
Our survey of the capacity of RNA and DNA virus infections to be transmitted, directly or indirectly, between humans leads to several conclusions and practical suggestions for improving surveillance of emerging infectious diseases and targeting efforts to identify future public health threats. In support of these conclusions, the World Health Organization recently published list of priority emerging infectious diseases and corresponding viruses (38) included 6 of the viruses in Table 2.
A major observation is that the taxonomic diversity of viruses that are possible threats to public health is wide, but bounded. Most human infective viruses are closely related to viruses of other mammals and some to viruses of birds. There are no indications that humans acquire new viruses from any other source. However, diversification within human populations occurs and is a prominent feature of some DNA virus taxa (e.g., family Papillomaviridae) (4).
In general, however, our knowledge of origins of human viruses is still incomplete. Although the origins of HIV-1 have been extensively investigated (16), for most other viruses, even level 4 viruses, little or no research has occurred. An origins initiative (9) would help establish the routes into human populations that have been used by other viruses.
Transmissibility within human populations is a key determinant of epidemic potential. Many viruses that can infect humans are not capable of being transmitted by humans; most human transmissible viruses that emerge already have that capability at first human infection or acquire it relatively rapidly. If transmission from humans would require a change in a phylogenetically conserved trait, such as tissue tropism or transmission route (4), then such viral paradigm shifts will probably be extremely rare (39).
Even when a virus is capable of transmission between humans, the critical threshold R0>1 is not always achieved. However, because changes in virus traits or host population characteristics can influence R0, level 3 viruses (Table 2) are of special interest from a public health perspective, and of special concern when, like MERS-CoV, they also cause severe illness. Demonstrating human transmissibility is often difficult, but essential. The best evidence is likely to come from virus genome sequencing studies. These studies should be a public health priority (40).
We currently have few clues to help us predict which mammalian or avian viruses might pose a threat to humans and, especially, which might be transmissible between humans. One argument in favor of experimental studies of these traits, including controversial gain of function experiments (30), is that they could help guide molecular surveillance for high-risk virus lineages in nonhuman reservoirs.
The first line of defense against emerging viruses is effective surveillance (40). A better understanding of which kinds of viruses in which circumstances pose the greatest risk to human health would enable evidence-based targeting of surveillance efforts, which would reduce costs and increase probable effectiveness of this endeavor.
Dr. Woolhouse is professor of Infectious Disease Epidemiology at the Centre for Immunity, Infection and Evolution at the University of Edinburgh, Edinburgh, UK. His primary research interests are pathogen emergence and antimicrobial drug resistance.
Acknowledgments
We thank David McCulloch, Cristina Moreno, and Matthew Hall for assistance during this study.
This study was supported by the Wellcome Trust Vietnam Initiative on Zoonotic Infections Project grant 093724/B/10/Z to M.E.J.W. and European Union 2020 grant COMPARE #643476. L.B. was supported by a Natural Environment Research Council PhD studentship, and S.L. was supported by a University of Edinburgh Chancellor’s Fellowship and the Biotechnology and Biological Sciences Research Council Strategic Programme grant BB/J004227/1.
References
- Taylor LH, Latham SM, Woolhouse ME. Risk factors for human disease emergence. Philos Trans R Soc Lond B Biol Sci. 2001;356:983–9.DOIPubMedGoogle Scholar
- Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Global trends in emerging infectious diseases. Nature. 2008;451:990–3.DOIPubMedGoogle Scholar
- Morse SS, Mazet JA, Woolhouse M, Parrish CR, Carroll D, Karesh WB, Prediction and prevention of the next pandemic zoonosis. Lancet. 2012;380:1956–65.DOIPubMedGoogle Scholar
- King AM, Lefkowitz E, Adams MJ, Carstens EB. Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Amsterdam: Elsevier; 2012.
- Woolhouse ME, Haydon DT, Antia R. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol Evol. 2005;20:238–44.DOIPubMedGoogle Scholar
- Anderson RM, May RM. Infectious diseases of humans: dynamics and control. New York: Oxford University Press; 1991.
- Hay SI, Battle KE, Pigott DM, Smith DL, Moyes CL, Bhatt S, Global mapping of infectious disease. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120250.DOIPubMedGoogle Scholar
- Woolhouse ME. Population biology of emerging and re-emerging pathogens. Trends Microbiol. 2002;10(Suppl):S3–7.DOIPubMedGoogle Scholar
- Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–83.DOIPubMedGoogle Scholar
- J Woolhouse ME, Adair K, Brierley L. RNA viruses: a case study of the biology of emerging infectious diseases. Microbiol Spectr. 2013;1:OH-0001–2012.DOIPubMedGoogle Scholar
- Woolhouse ME, Adair K. The diversity of human RNA viruses. Future Virol. 2013;8:159–71 .DOIGoogle Scholar
- Kuiken T, Holmes EC, McCauley J, Rimmelzwaan GF, Williams CS, Grenfell BT. Host species barriers to influenza virus infections. Science. 2006;312:394–7.DOIPubMedGoogle Scholar
- Davies TJ, Pedersen AB. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc Biol Sci. 2008;275:1695–701.DOIPubMedGoogle Scholar
- Cooper N, Nunn CL. 2013 Identifying future zoonotic disease threats. Evol Med Public Health. 2013;1:27–36.DOIPubMedGoogle Scholar
- Khabbaz RF, Heneine W, George JR, Parekh B, Rowe T, Woods T, Brief report: infection of a laboratory worker with simian immunodeficiency virus. N Engl J Med. 1994;330:172–7.DOIPubMedGoogle Scholar
- Sharp PM, Hahn BH. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med. 2011;1:a006841.DOIPubMedGoogle Scholar
- Chowell G, Hengartner NW, Castillo-Chavez C, Fenimore PW, Hyman JM. The basic reproductive number of Ebola and the effects of public health measures: the cases of Congo and Uganda. J Theor Biol. 2004;229:119–26.DOIPubMedGoogle Scholar
- Schwartz O, Albert ML. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol. 2010;8:491–500.DOIPubMedGoogle Scholar
- Fauci AS, Morens DM. Zika virus in the Americas: yet another arbovirus threat. N Engl J Med. 2016;374:601–4.DOIPubMedGoogle Scholar
- Blumberg S, Lloyd-Smith JO. Inference of R(0) and transmission heterogeneity from the size distribution of stuttering chains. PLoS Comput Biol. 2013;9:e1002993.DOIPubMedGoogle Scholar
- Lord CC, Barnard B, Day K, Hargrove JW, McNamara JJ, Paul RE, Aggregation and distribution of strains in microparasites. Philos Trans R Soc Lond B Biol Sci. 1999;354:799–807.DOIPubMedGoogle Scholar
- Yates A, Antia R, Regoes RR. How do pathogen evolution and host heterogeneity interact in disease emergence? Proc Biol Sci. 2006;273:3075–83.DOIPubMedGoogle Scholar
- Jansen VA, Stollenwerk N, Jensen HJ, Ramsay ME, Edmunds WJ, Rhodes CJ. Measles outbreaks in a population with declining vaccine uptake. Science. 2003;301:804.DOIPubMedGoogle Scholar
- Woolhouse ME, Gaunt E. Ecological origins of novel human pathogens. In: Relman DA, Hamburg MA, Choffnes ER, Mack A, editors. Microbial evolution and co-adaptation, Washington (DC): National Academies Press; 2009. p. 208–29.
- Breban R, Riou J, Fontanet A. Interhuman transmissibility of Middle East respiratory syndrome coronavirus: estimation of pandemic risk. Lancet. 2013;382:694–9.DOIPubMedGoogle Scholar
- Fine PE, Jezek Z, Grab B, Dixon H. The transmission potential of monkeypox virus in human populations. Int J Epidemiol. 1988;17:643–50.DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Outbreaks chronology: Ebola virus disease [cited 2015 Feb 1]. http://www.cdc.gov/vhf/ebola/outbreaks/history/chronology.html#modalIdString_outbreaks
- Woolhouse M, Antia R. Emergence of new infectious diseases. In: Stearns SC, Koella JK, editors. Evolution in health and disease. 2nd ed. Oxford (UK): Oxford University Press; 2008. p. 215–28.
- Domingo E, Martínez-Salas E, Sobrino F, de la Torre JC, Portela A, Ortín J, The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance—a review. Gene. 1985;40:1–8.DOIPubMedGoogle Scholar
- Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Airborne transmission of influenza A/H5N1 virus between ferrets. Science. 2012;336:1534–41.DOIPubMedGoogle Scholar
- Arinaminpathy N, McLean AR. Evolution and emergence of novel human infections. Proc Biol Sci. 2009;276:3937–43.DOIPubMedGoogle Scholar
- Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. MBio. 2014;5:e01062–13.DOIPubMedGoogle Scholar
- Gire SK, Goba A, Andersen KG, Sealfon RS, Park DJ, Kanneh L, Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–72.DOIPubMedGoogle Scholar
- Jombart T, Cori A, Didelot X, Cauchemez S, Fraser C, Ferguson N. Bayesian reconstruction of disease outbreaks by combining epidemiologic and genomic data. PLoS Comput Biol. 2014;10:e1003457.DOIPubMedGoogle Scholar
- Volz EM, Kosakovsky Pond SL, Ward MJ, Leigh Brown AJ, Frost SD. Phylodynamics of infectious disease epidemics. Genetics. 2009;183:1421–30.DOIPubMedGoogle Scholar
- Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–97.DOIPubMedGoogle Scholar
- Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature. 2013;502:241–4.DOIPubMedGoogle Scholar
- World Health Organization. Blueprint for R&D preparedness and response to public health emergencies due to highly infectious pathogens, 2015 [cited 2016 Aug 21]. http://www.who.int/csr/research-and-development/meeting-report-prioritization.pdf?ua=1
- Belshaw R, Gardner A, Rambaut A, Pybus OG. Pacing a small cage: mutation and RNA viruses. Trends Ecol Evol. 2008;23:188–93.DOIPubMedGoogle Scholar
- Woolhouse ME, Rambaut A, Kellam P. Lessons from Ebola: Improving infectious disease surveillance to inform outbreak management. Sci Transl Med. 2015;7:307rv5.DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 22, Number 12—December 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Mark E.J. Woolhouse, Centre for Immunity, Infection, and Evolution, University of Edinburgh, Ashworth Laboratories, Charlotte Auerbach Rd, Edinburgh EH9 3FL, UK
Top