Volume 25, Number 1—January 2019
Research
Variable Protease-Sensitive Prionopathy Transmission to Bank Voles
Cite This Article
Citation for Media
Abstract
Variably protease-sensitive prionopathy (VPSPr), a recently described human sporadic prion disease, features a protease-resistant, disease-related prion protein (resPrPD) displaying 5 fragments reminiscent of Gerstmann-Sträussler-Scheinker disease. Experimental VPSPr transmission to human PrP–expressing transgenic mice, although replication of the VPSPr resPrPD profile succeeded, has been incomplete because of second passage failure. We bioassayed VPSPr in bank voles, which are susceptible to human prion strains. Transmission was complete; first-passage attack rates were 5%–35%, and second-passage rates reached 100% and survival times were 50% shorter. We observed 3 distinct phenotypes and resPrPD profiles; 2 imitated sporadic Creutzfeldt-Jakob disease resPrPD, and 1 resembled Gerstmann-Sträussler-Scheinker disease resPrPD. The first 2 phenotypes may be related to the presence of minor PrPD components in VPSPr. Full VPSPr transmission confirms permissiveness of bank voles to human prions and suggests that bank vole PrP may efficiently reveal an underrepresented native strain but does not replicate the complex VPSPr PrPD profile.
Sporadic prion diseases are classified according to phenotype as well as the pairing of the prion protein (PrP) genotype at the methionine (M)/valine (V) polymorphic codon 129 and the conformational characteristics of the abnormal or disease-associated PrP (PrPD). These characteristics include electrophoretic mobility and the ratio of the PrPD fragments that are resistant to proteinase K (PK) digestion (Appendix Table 1) (1). According to these criteria, the 3 major types of sporadic prion disease are sporadic Creutzfeldt-Jakob disease (sCJD), sporadic fatal insomnia, and variably protease-sensitive prionopathy (VPSPr) (2–5).
VPSPr was first reported in 2008 and further defined in 2010 (6–8) as a sporadic prion disease distinct from sCJD. Since then, 37 cases have been reported, consistent with a prevalence rate of 1%–2% for all sporadic prion diseases (8). Similar to sCJD, VPSPr targets all 3 PrP genotypes. However, the prevalence of the 3 genotypes at codon 129 (MM, MV, and VV) greatly differs, indeed is almost inverted, in the 2 diseases: homozygosity VV is the most common (65%) genotype in VPSPr and the least common (16%) in sCJD (2,9). Furthermore, at variance with sCJD, in which the 129 genotype is a determinant of disease phenotype and PrPD characteristics, the 129 genotype influence on phenotype, although present, is subtle (3,7,8). These differences point to a distinct role of the 129 genotype as a risk factor and imply that the etiologic-pathogenetic mechanisms of the 2 diseases differ.
Although the histopathology of VPSPr is distinct (e.g., spongiform degeneration, frequent presence of PrP microplaques, and a recognizable PrPD immunostaining pattern), the hallmarks of VPSPr are the characteristics of its PrPD. In contrast to virtually all other sporadic human prion diseases, in which PK-resistant PrPD (resPrPD) electrophoretically separates into 3 major bands, VPSPr resPrPD characteristically separates into 5 bands. Furthermore, although the 3 bands of resPrPD are all cleaved by PK exclusively at the N terminus and separate according to the presence of 2, 1, or 0 sugar moieties, VPSPr resPrPD bands include only the monoglycosylated and unglycosylated forms, which are cleaved either only at the N terminus or at both the N- and C-termini. Thus, the C-terminus–truncated resPrPD lacks the GPI (glycosylphosphatidylinositol) anchor. Additional variances concerning immunoreactivity characteristics, ratios of PK-resistant and PK-sensitive PrPD species, and conformational properties including aggregate size, have also been observed (6–8). These distinctive properties point to VPSPr PrPD as a prion strain different from those of other sporadic prion diseases. However, the VPSPr prion shares the multiplicity of the resPrPD electrophoretic bands with prions from a subset of inherited prion diseases referred to as Gerstmann-Sträussler-Scheinker disease (GSS), prompting the suggestion that VPSPr is the sporadic form of GSS (7,10). Furthermore, the presence of small amounts of sCJD-like 3-band resPrPD has also been signaled in VPSPr (6,11,12).
Disease transmission to receptive hosts is a valuable way to further define the characteristics of strains associated with prion diseases. VPSPr has been experimentally transmitted to 3 lines of transgenic mice expressing normal PrP or cellular human PrP (PrPC), harboring residue M, V, or MV at residue 129 (13,14). Data in all experiments were essentially similar. Inoculated mice remained asymptomatic, but half showed focal PrPD plaques with minimal spongiform degeneration, and PrPD mimicking the electrophoretic profile of the native PrPD on immunoblot was demonstrated in about one third of the inoculated mice. No transmission was observed at second passage.
The bank vole, a small rodent resembling the mouse with which it shares the entire sequence of normal PrP or PrPC except for 8 aa, but whose sequence differs from human PrPC by 15 aa, has recently emerged as a particularly permissive host. Bank voles and transgenic mice expressing bank vole PrPC have been successfully infected after challenge with human and animal prion diseases that are hard to transmit even to recipients expressing homologous PrPC (15–18).
We studied transmission of VPSPr from patients with MM, MV, and VV codon 129 genotypes to bank voles harboring either the PrP genotype 109M (bv109M) or 109I (bv109I). Although the attack rate was generally low at first passage, it consistently raised to 100% at second passage, when survival times also decreased on average by >50%. We identified 3 PrPD isoforms with the characteristics of distinct strains in the affected bank voles.
The inocula used in the first passage were brain homogenates from 7 persons with a definitive diagnosis of VPSPr: 2 with genotypes 129MM, 3 with 129MV, and 2 with 129VV. Homogenate was inoculated into the cerebrum of 205 bank voles according to previously described procedures (16). The bank vole brains were processed for histopathology, immunohistochemistry, lesion profiles, and paraffin-embedded tissue (PET) blots according to previously reported procedures (15). Western blot was performed according to Notari et al. (19). The insoluble fraction was prepared according to previously described procedures (20). Preparation of monoclonal antibodies is described in the Appendix. Statistical significance was determined by 1-way analysis of variance, followed by the Tukey multiple comparison test.
Transmission Characteristics
At first passage, attack rates of VPSPr were 35% (29/82) in bv109I and 5% (3/59) in bv109M (Table 1; Appendix Table 2). The 2 bank vole genotypes diverged as to disease transmission in 2 ways. First, all VPSPr 129 genotypes were transmitted to bv109I, but bv109M were not susceptible to VPSPr-VV. Second, bv109I propagated 3 distinct histopathologic phenotypes and matching PrPD types (hereafter identified as T1, T2, and T3), but bv109M replicated the T1 phenotype exclusively. A more detailed analysis in bv109I, although limited by the low number of animals in each subset, suggested a lower attack rate for VPSPr-VV, the most common form of human VPSPr, compared with the MM and MV genotypes and a prevalence for T3 that was 11% higher than that for T1 and 106% higher than that for T2 (Table 2). Overall survival times were 575 days postinoculation (dpi) for bv109I and 411 dpi for bv109M. However, when we considered only the bank voles associated with the T1 phenotype, because bv109M were exclusively associated with T1, the dpi difference became smaller: 490 dpi for bv109I and 411 dpi for bv109M (Tables 1, 2). As for survival times related to T1–T3 phenotypes and VPSPr genotypes, the survival times for T2 were nearly twice those for T1 and T3 (Table 2).
Second passage in bv109I was invariably characterized by a 100% attack rate, a 40%–61% decrease in survival times, and conservation of the original phenotype (Table 1). A similar trend was observed for bv109M.
Histopathology and Immunohistochemistry
Figure 1
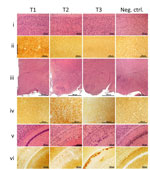
Figure 1. Histopathology and prion protein (PrP) immunohistochemistry images of brain regions from variably protease-sensitive prionopathy (VPSPr)–inoculated bank voles 109I harboring the histopathologic phenotypes T1, T2, or T3. For T1 bank voles, the...
Figure 2

Figure 2. Profiles of topographic distribution and severity of spongiform degeneration in the brains of bank voles harboring T1–T3 phenotypes after inoculation with brain homogenate from variably protease sensitive prionopathy (VPSPr) and control...
Figure 3
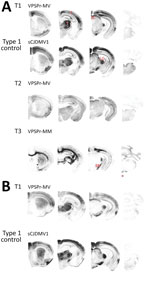
Figure 3. Representative paraffin-embedded tissue (PET) blots of protease-resistant, disease-related prion protein (resPrPD) distribution in phenotypes T1–T3 and controls. A) For T1, PrPD predominated in cerebral cortex (C), thalamus (T), superior colliculus (SC),...
Phenotype T1 featured finely vacuolated spongiform degeneration often involving the entire thickness of the neocortex, including the molecular layer but occasionally also showing a laminar distribution (Figure 1). On second passage, the spongiform degeneration appeared to be more widespread, also affecting the hippocampus and subcortical structures such as basal nuclei, thalamus, and superior colliculi but not the cerebellum. PrP immunohistochemistry demonstrated punctate deposits often co-distributed with spongiform degeneration (Figure 1, column T1, row ii). At second passage, T1 features did not differ significantly between bv109M and bv109I. Furthermore, T1 also resembled the histopathologic phenotype shown by bv109M and bv109I after inoculation with sCJDMM1 or sCJDMV1, respectively (Figures 2, 3; Appendix Figure 1)
In phenotype T2, spongiform degeneration affected predominantly subcortical structures over neocortical regions, especially the hypothalamus with the apparent exclusion of the mammillary bodies (Figure 1, column T2, row iii). PrP immunohistochemistry showed granular deposits occasionally resembling miniature plaquelike formations rather than the punctate deposits of the T1 phenotype (Figure 1, column T2, row iv).
Phenotype T3 was characterized by the paucity of spongiform degeneration in the cerebral neocortex and subcortical gray matter structures; spongiform degeneration was often prominent in the regions of the hemispheric white matter lying above the hippocampus and in the corpus callosum, where parenchyma was occasionally disorganized with glial reaction. PrP immunostaining was mostly limited to those regions where it often aggregated in confluent plaque-like deposits but not well-formed plaques (Figure 1, column T3, row v, and column Tc, row vi). No remarkable differences were detected between first and second passages. Overall, the T3 histopathologic phenotype resembled that shown by bv109I after inoculation with brain homogenates from some GSS subtypes (16).
It is noteworthy that the T1–T3 phenotypes were never observed to coexist in 1 animal, although distinct phenotypes were often observed in bank voles receiving the same inoculum. Although all 3 phenotypes occurred after inoculation with VPSPr-MM or -MV, the sole phenotype associated with VPSPr-VV inoculation was T3 (Table 1).
Lesion Profiles and PET Blots
Profiles of spongiform degeneration as a function of lesion severity and brain anatomic location confirmed the distinctive characteristics of the T1–T3 phenotypes (Figure 2, panel A; Appendix Figure 1). The T1 spongiform degeneration profile in bv109I did not differ significantly from that of bv109M; both mirrored the profiles of bv109I inoculated with sCJDMV1 and bv109M inoculated with sCJDMM1 brain homogenate (Figure 2, panel B; Appendix Figure 1).
The PET blot patterns of brain PrPD distribution were also quite distinct in the 3 phenotypes and, overall, reproduced the spongiform degeneration distribution (Figures 2, 3). In T1, PrPD was well represented in selected regions including cerebral neocortex and hippocampus, basal nuclei, thalamus, superior colliculi, geniculate nuclei, and substantia nigra but not in the cerebellum and lower brain stem. No significant variations were detected between PrPD distributions at first and second passages (data not shown). PrPD distributions were also similar in bv109I and bv109M inoculated with classic sCJDMV1 and sCJDMM1 prions, respectively (Figure 3, panel B). In the T2 phenotype, PrPD appeared to be present in moderate and uniform amounts in several anatomic regions such as neocortex and hippocampus, thalamus, and superior colliculi (Figure 3). The T3 phenotype was characterized by the striking presence of PrPD in hippocampus and white matter structures (Figure 3).
PrPD Characterization
Figure 4

Figure 4. Immunoblot characteristics of protease-resistant, disease-related prion protein (resPrPD) distribution in phenotypes T1–T3 and controls. Regular and long exposures revealed the overall similarity of the 3-band profiles in T1 and T2, but...
Figure 5

Figure 5. Glycoform ratio of protease-resistant, disease-related prion protein (resPrPD) in phenotypes T1 and T2. The ratio of resPrPD associated with T1 (T1 109I) was 48% for diglycosylated, 44% for monoglycosylated, and 8%...
Immunoblot analysis confirmed the presence of 3 distinct resPrPD electrophoretic profiles that matched the 3 histopathologic phenotypes. When probed with antibodies 9A2 and 12B2, resPrPD associated with the T1 phenotype populated 3 bands of ≈32, 26, and 21 kDa, representing the 3 resPrPD glycoforms, and by a fragment of 7 kDa (Figure 4). An additional C-terminal fragment of ≈13 kDa, possibly homologous to the human C-terminal fragment 12/13 (20), was detected by the C-terminal antibody SAF84 (Figure 4). Glycoform ratios showed a comparable representation of the diglycosylated and monoglycosylated forms of resPrPD (Figure 5; Appendix Figure 2). The electrophoretic profile and glycoform ratios of resPrPD T1 conformer were indistinguishable from those of resPrPD observed in bank voles inoculated with sCJDMM1 or sCJDMV1 prions, used as controls for human resPrPD type 1 in bank voles (Figures 4, 5; Appendix Figure 2; data not shown).
The resPrPD profile associated with the T2 phenotype showed 3 bands of ≈30, 24, and 19 kDa (i.e., all that had an ≈2-kDa faster electrophoretic mobility than the corresponding bands of resPrPD T1) (Figure 4). The 7-kDa fragment was not detected in T2 (Figure 4). In contrast to T1, the T2 glycoform ratio was characterized by the unambiguous predominance of the monoglycosylated component (Figure 5). In summary, bank vole resPrPD T2 differed from the T1 conformer by overall 2-kDa faster mobility, the absence of the 7-kDa fragment, and marked predominance of the monoglycoform. The striking feature of the resPrPD associated with the T3 phenotype was the predominant presence of the 7-kDa fragment detected by 9A2 and 12B2 but not by SAF84, demonstrating its internal origin and the absence of glycosylation sites (Figure 4).
Figure 6

Figure 6. Relative quantities of totPrPD and resPrPD in T1–T3 phenotypes. totPrPD accounted for 93.1% and resPrPD for 81.3% of total PrP recovered from bank voles harboring the T1 phenotype. Corresponding percentages for...
Additional divergent features emerged when amounts of totPrPD (i.e., PK-sensitive plus resPrPD fractions) were assessed as percentages of total PrP, comprising PrPC and totPrPD (Figure 6). A significantly larger component of totPrPD was resPrPD in T1 than in T2 (81% vs. 33%); totPrPD fractions were similar (93% for T1, 91% for T2). T3 differed significantly: totPrPD accounted for 8% and resPrPD accounted for 0.2% of total PrP (Figure 6; Appendix Figure 3).
The permissiveness of bank vole PrPC is well known (15,16,18,21–27); it is exemplified by the observation that, despite the mere 8-aa PrPC divergence between bank voles and mice, a variety of human and animal prion diseases not transmissible to mice are infectious to bank voles and transgenic mice expressing bank vole PrP (15,16,18,22,24). Conversely, the 15-aa difference from the human PrPC primary sequence does not impede the efficient transmission of a wealth of sporadic and inherited human prion diseases (15,16). This striking permissiveness has been attributed to the presence of several asparagine and glutamine residues in and around the β2–α2 loop that would result in a PrPC conformation compatible with the conformations of a large number of PrPD strains (21). Furthermore, the polymorphism at bank vole codon 109 adds further complexity to the interaction with exogenous strains (18,28).
We undertook systematic transmission of VPSPr brain homogenates to bv109M and bv109I after failure to consistently transmit VPSPr to humanized transgenic mice. Overall, transmission was favored by the 109I genotype, which propagated all 3 VPSPr 129 genotypes while bv109M failed to transmit VPSPr-VV. However, at first passage in bv109I, the mean attack rate (35%) was fairly low and the mean survival time (575 dpi) quite extended (Table 1). These conditions changed at second passage, when the attack rate became 100% in all transmission experiments and survival times decreased on average by 53% (Table 1). These findings point to the existence of a substantial barrier at first passage, which, judging from the 100% attack rate, is probably largely diminished or vanished at second passage. In view of the aforementioned easy transmissibility of other human prion diseases, the barrier appears to be conformational rather than caused by species-related variations in amino acid sequence of PrPC (15,16); the barrier might be associated with the misfolding of VPSPr PrPD, which may be peculiar because after PK digestion it results in an array of highly heterogeneous fragments and apparently the failure to convert one of the glycoforms (6,7). Similarly, the clear effect of the genotype at codon 129 on the attack rate (which was 3–4 times lower for bank voles inoculated with VPSPr-VV prions compared with VPSPr-MM and -MV), along with the lack of transmission of VPSPr-VV to bv109M but not to bv109I, points to conformational differences between PrPD species associated with the 129 genotypes in VPSPr (16). This notion is further supported by previous data showing higher PK sensitivity (7) and conformational stability of PrPD (29) in VPSPr-VV compared with VPSPr-MM and -MV.
The comparative study of VPSPr bioassay in bank voles and humanized transgenic mice revealed substantial differences. VPSPr-challenged mice invariably remained asymptomatic, and all histologically positive mice failed to transmit at second passage. Furthermore, the VPSPr-MV subtype was never transmitted to mice 129M or 129V, and the general attack rate (assessed histopathologically) was low (54%); resPrPD was demonstrated in only 34% of the challenged mice despite the 2–8 times normal levels of PrP expression for most mice (13). However, in contrast to bank voles, positive mice generated a resPrPD conformer very similar to that of VPSPr for electrophoretic profile, glycosylation pattern, and antibody immunoreactivity, although it exhibited higher protease resistance.
Data from a previous study of transmission to humanized transgenic mice and bv109M of an sCJDMV variant with an atypical glycoform profile (CJD-MVAG) partially resembled ours (17). Challenged transgenic mice remained asymptomatic and negative at neuropathologic examination, but 22% of them reproduced the original resPrPD electrophoretic profile and glycotype of the inoculum. In contrast to humanized transgenic mice, bank voles had full-blown disease develop featuring 3, although partially merging, histopathologic phenotypes along with 3 distinct resPrPD conformers, none of which mimicked the profile and glycotype of the inoculum (17). Remarkably, the glycoform variation of sCJDMVAG resembles that of VPSPr because both resPrPD species lack the diglycosylated isoform, implicating this variation as one of the possible causes of bank vole failure to accurately replicate exogenous PrPD (17).
Three subtypes of GSS (which VPSPr resembles in terms of the ladder-like electrophoretic profile and the sensitivity to PK of resPrPD) have also recently been transmitted to bank voles and 1 GSS subtype to humanized transgenic mice (16,30). Despite the well-known difficulty of transmitting GSS to rodents, bank voles challenged with 2 major GSS subtypes associated with PrP mutations A117V and F198S (GSSA117V, GSSF198S) showed no evidence of species or mutation barrier. Transmission was comparatively more difficult with the third GSSP102L subtype, in which resPrPD displays 2 sets of fragments: either the 8-kDa fragment associated with the 30–21 kDa glycoform triplet (31,32) or the 8-kDa fragment alone. After inoculation, the 2-fragment set was never replicated, and the ≈8-kDa fragment alone occasionally was inaccurately reproduced as a 7-kDa fragment (16,28). To date, only GSSA117V has been transmitted to 2 lines of transgenic mice expressing human PrPD harboring the A117V transition (30). Although transmission features diverged in the 2 lines, both seemed to reproduce the 7-kDa fragment that is the only strongly resPrPD fragment in this disease.
Combined, these experiments indicate that PrPC characteristics, and possibly other host factors (25), enable bank voles to be more permissive hosts (despite the species barrier) than transgenic mice expressing conspecific PrPC, confirming the empirical aspect of the species barrier. However, bank vole PrPC can hardly reproduce faithfully complex features of human atypical prion isolates, a task that may require PrPC from the same species.
A remarkable finding of this study is the occurrence of 3 well-defined histopathologic phenotypes (T1–T3), which displayed discrete PrPD brain distribution and were linked to PrPD conformers easily distinguishable by electrophoretic profile and glycosylation characteristics. The 3 phenotypes also differed by mean survival times at first and second passages. Remarkably, the T1–T3 phenotypes were often generated by the same inoculum but never co-occurred in the same bank vole. Combined, these features define the T1–T3 PrPD conformers as distinct strains, raising the issue of their origin. Both histopathologic and resPrPD electrophoretic characteristics of the T1 phenotype are essentially indistinguishable from those of bank voles inoculated with sCJDMV1. Data on transmission of sCJDMM2, available only for bv109M, show that the electrophoretic profile of the newly formed resPrPD matches the T2 resPrPD of this study (15). Although the T1 and T2 representations of totPrPD and resPrPD are not known in bank voles inoculated with sCJDMM1 and sCJDMM2 prions, the values we observed after VPSPr inoculation are comparable to those reported for the original sCJD, in which totPrPD and resPrPD reportedly accounted for 53.5% and 48.2% of total PrP in sCJDMM1 (6; L. Cracco et al., unpub. data). Therefore, transmission to bank voles suggests that VPSPr PrPD T1 and T2 are related to human PrPD types 1 and 2, respectively. In contrast, phenotype T3 is the most divergent, especially for spongiform degeneration and PrPD deposition, mostly limited to white matter regions, and electrophoretic profile, where resPrPD recovered as a band of 7 kDa, was the major component shared with the complex pattern of VPSPr resPrPD. The T3 histopathologic phenotypes including the PrP immunostaining pattern matched also the bank vole phenotype of GSSA117V and GSSP102L associated with the 8-kDa fragment only (16). The exceedingly low representation of the totPrPD and resPrPD components of total PrP in T3 is reminiscent of the corresponding data reported in VPSPr-VV, in which totPrPD accounted for 3.4% and resPrPD for 0.83% of total PrP (6). The marked underrepresentation of totPrPD and resPrPD in T3 is especially puzzling considering that attack rate and survival time are not very different from those of T2 and T1, respectively. The apparent relative high efficiency of T3 might be explained by the high representation of oligomers (33). Alternatively, the T3 underrepresentation of totPrPD relative to total PrP might reflect the lack of PrPC down-regulation by T3 compared with T1 and T2, which would result in the relative increase of the total PrP pool (33,34).
A mechanism put forward for the lack of fidelity in cross-species transmission of the prion strain (25,35–37) is based on evidence that the dominant strain is selected from an array of strains that persist as substrains. In cross-species transmissions, substrains may be selected over the dominant strain (38–40). In the context of VPSPr, this mechanism is particularly intriguing, given that small quantities of PrPD conformers with electrophoretic mobilities similar to those of human PrPD types 1 were originally observed in a few cases by Gambetti et al (6); more recently, the presence of PrPD type 2 in VPSPr, mostly in subcortical nuclei and in cerebellum, has been reported (11,12). These 2 components would be propagated faithfully in T1 and T2, and T3, which consistently shares only the 7-kDa fragment with the VPSPr resPrPD, might represent the isolation of this GSS-like component, of VPSPr resPrPD or the unsuccessful attempt to fully reproduce the dominant strain associated with this disease. We and others have occasionally observed an underrepresented 7-kDa fragment in sCJDMM1 (41; S. Notari, P. Gambetti, P. Parchi, unpub. data). Thus, it is tempting to speculate that the 7-kDa fragment observed in bank voles inoculated with sCJDMM1 and sCJDMV1 prions is related to the presence and possibly the infectivity of such fragment in the sCJDMM(MV)1 subtype.
In conclusion, on the basis of the first full transmission of VPSPr, our study confirms the permissiveness of bank voles to human prion diseases and suggests that bank voles are competent to reveal minor strain variants in prion diseases, such as resPrPD types 1 and 2 reported in VPSPr and, possibly, the ≈7-kDa fragment observed in sCJDMM1 and sCJDMV1. However, our study also underscores the limited competence of bank vole PrPC to faithfully reproduce the multiband profile of VPSPr resPrPD that probably reflects the complex conformation of the prion seed in this disease.
Dr. Nonno is a research scientist in the Emerging Zoonosis Unit at the Istituto Superiore di Sanità in Rome, Italy; his primary research interests include prion strain characterization and the zoonotic potential of animal prion diseases. Dr. Notari is an instructor in the Department of Pathology at Case Western Reserve University, Cleveland, Ohio, USA; his research focuses mainly on prion diseases, particularly prion molecular characteristics and infectivity.
Acknowledgments
We thank Janis Blevins, Katie Eppich, Yvonne Cohen, and the other personnel of the National Prion Disease Pathology Surveillance Center as well as Geraldina Riccardi, Stefano Marcon, Paolo Frassanito, and Marcello Rossi for their skillful assistance provided during the course of this study.
This work was supported by the National Institutes of Health (P01AI106705, 5R01NS083687) and the Charles S. Britton Fund (to P.G.).
References
- Gambetti P. Creationism and evolutionism in prions. Am J Pathol. 2013;182:623–7. DOIPubMedGoogle Scholar
- Gambetti P, Cali I, Notari S, Kong Q, Zou WQ, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121:79–90. DOIPubMedGoogle Scholar
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11:618–28. DOIPubMedGoogle Scholar
- Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol. 2011;121:91–112. DOIPubMedGoogle Scholar
- Poggiolini I, Saverioni D, Parchi P. Prion protein misfolding, strains, and neurotoxicity: an update from studies on mammalian prions. Int J Cell Biol. 2013;2013:910314.
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. DOIPubMedGoogle Scholar
- Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68:162–72. DOIPubMedGoogle Scholar
- Notari S, Appleby B, Gambetti P. Variably protease sensitive prionopathy. In: Pocchiari M, Manson J. editors. Handbook of clinical neurology, vol. 153. San Diego: Elsevier; 2018. p. 175–190.
- Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278–87. DOIPubMedGoogle Scholar
- Pirisinu L, Nonno R, Esposito E, Benestad SL, Gambetti P, Agrimi U, et al. Small ruminant nor98 prions share biochemical features with human gerstmann-sträussler-scheinker disease and variably protease-sensitive prionopathy. PLoS One. 2013;8:e66405. DOIPubMedGoogle Scholar
- Rodríguez-Martínez AB, López de Munain A, Ferrer I, Zarranz JJ, Atarés B, Villagra NT, et al. Coexistence of protease sensitive and resistant prion protein in 129VV homozygous sporadic Creutzfeldt-Jakob disease: a case report. J Med Case Reports. 2012;6:348. DOIPubMedGoogle Scholar
- Peden AH, Sarode DP, Mulholland CR, Barria MA, Ritchie DL, Ironside JW, et al. The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol Commun. 2014;2:152. DOIPubMedGoogle Scholar
- Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P, et al. Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis. 2014;20:2006–14. DOIPubMedGoogle Scholar
- Diack AB, Ritchie DL, Peden AH, Brown D, Boyle A, Morabito L, et al. Variably protease-sensitive prionopathy, a unique prion variant with inefficient transmission properties. Emerg Infect Dis. 2014;20:1969–79. DOIPubMedGoogle Scholar
- Nonno R, Angelo Di Bari M, Agrimi U, Pirisinu L. Transmissibility of Gerstmann-Sträussler-Scheinker syndrome in rodent models: New insights into the molecular underpinnings of prion infectivity. Prion. 2016;10:421–33. DOIPubMedGoogle Scholar
- Pirisinu L, Di Bari MA, D’Agostino C, Marcon S, Riccardi G, Poleggi A, et al. Gerstmann-Sträussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep. 2016;6:20443. DOIPubMedGoogle Scholar
- Galeno R, Di Bari MA, Nonno R, Cardone F, Sbriccoli M, Graziano S, et al. Prion strain characterization of a novel subtype of Creutzfeldt-Jakob disease. J Virol. 2017;91:e02390–16. DOIPubMedGoogle Scholar
- Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014;10:e1003990. DOIPubMedGoogle Scholar
- Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P, et al. A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest. 2007;87:1103–12. DOIPubMedGoogle Scholar
- Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem. 2003;278:40429–36. DOIPubMedGoogle Scholar
- Agrimi U, Nonno R, Dell’Omo G, Di Bari MA, Conte M, Chiappini B, et al. Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog. 2008;4:e1000113. DOIPubMedGoogle Scholar
- Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol. 2008;89:2975–85. DOIPubMedGoogle Scholar
- Cosseddu GM, Nonno R, Vaccari G, Bucalossi C, Fernandez-Borges N, Di Bari MA, et al. Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 2011;7:e1002370. DOIPubMedGoogle Scholar
- Di Bari MA, Nonno R, Castilla J, D’Agostino C, Pirisinu L, Riccardi G, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 2013;9:e1003219. DOIPubMedGoogle Scholar
- Espinosa JC, Nonno R, Di Bari M, Aguilar-Calvo P, Pirisinu L, Fernández-Borges N, et al. PrPC governs susceptibility to prion strains in bank vole, while other host factors modulate strain features. J Virol. 2016;90:10660–9. DOIPubMedGoogle Scholar
- Orrú CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, et al. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog. 2015;11:e1004983. DOIPubMedGoogle Scholar
- Fernández-Borges N, Eraña H, Castilla J. Behind the potential evolution towards prion resistant species. Prion. 2018;12:83–7. DOIPubMedGoogle Scholar
- Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2:e12. DOIPubMedGoogle Scholar
- Pirisinu L, Marcon S, Di Bari MA, D’Agostino C, Agrimi U, Nonno R. Biochemical characterization of prion strains in bank voles. Pathogens. 2013;2:446–56. DOIPubMedGoogle Scholar
- Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A, et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog. 2013;9:e1003643. DOIPubMedGoogle Scholar
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci U S A. 1998;95:8322–7. DOIPubMedGoogle Scholar
- Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A. 2007;104:4712–7. DOIPubMedGoogle Scholar
- Mays CE, Kim C, Haldiman T, van der Merwe J, Lau A, Yang J, et al. Prion disease tempo determined by host-dependent substrate reduction. J Clin Invest. 2014;124:847–58. DOIPubMedGoogle Scholar
- Mays CE, van der Merwe J, Kim C, Haldiman T, McKenzie D, Safar JG, et al. Prion infectivity plateaus and conversion to symptomatic disease originate from falling precursor levels and increased levels of oligomeric PrPSc species. J Virol. 2015;89:12418–26. DOIPubMedGoogle Scholar
- Weissmann C. Mutation and selection of prions. PLoS Pathog. 2012;8:e1002582. DOIPubMedGoogle Scholar
- Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV. New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol. 2016;186:1006–14. DOIPubMedGoogle Scholar
- Moreno JA, Telling GC. Insights into mechanisms of transmission and pathogenesis from transgenic mouse models of prion diseases. Methods Mol Biol. 2017;1658:219–52. DOIPubMedGoogle Scholar
- Weissmann C, Li J, Mahal SP, Browning S. Prions on the move. EMBO Rep. 2011;12:1109–17. DOIPubMedGoogle Scholar
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. DOIPubMedGoogle Scholar
- Li J, Mahal SP, Demczyk CA, Weissmann C. Mutability of prions. EMBO Rep. 2011;12:1243–50. DOIPubMedGoogle Scholar
- Benedetti D, Fiorini M, Cracco L, Ferrari S, Capucci L, Brocchi E, et al. Molecular characterization of low molecular mass C-terminal fragments in different Creutzfeldt-Jakob disease subtypes. Presented at [Madrid, Spain.]. Prion. 2008;2008:8–10.
Figures
Tables
Cite This ArticleOriginal Publication Date: November 27, 2018
1These authors contributed equally to this article.
Table of Contents – Volume 25, Number 1—January 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Pierluigi Gambetti, Case Western Reserve University, 2085 Adelbert Rd, Rm 419, Cleveland, OH 44160, USA; ; Umberto Agrimi, Istituto Superiore di Sanità, Viale Regina Elena 299, Rome 00161, Italy
Top