Foodborne Origin and Local and Global Spread of Staphylococcus saprophyticus Causing Human Urinary Tract Infections
Opeyemi U. Lawal, Maria J. Fraqueza, Ons Bouchami, Peder Worning, Mette D. Bartels, Maria L. Gonçalves, Paulo Paixão, Elsa Gonçalves, Cristina Toscano, Joanna Empel, Małgorzata Urbaś, M. Angeles Domínguez, Henrik Westh, Hermínia de Lencastre, and Maria Miragaia

Author affiliations: Universidade Nova de Lisboa, Oeiras, Portugal (O.U. Lawal, O. Bouchami, H. de Lencastre, M. Miragaia); Centre for Interdisciplinary Research in Animal Health (CIISA), Universidade de Lisboa, Lisbon, Portugal (M.J., Fraqueza); Hvidovre University Hospital, Hvidovre, Denmark (P. Worning, M.D. Bartels, H. Westh); SAMS Hospital, Lisbon (M.L. Gonçalves); Hospital da Luz, Lisbon (P. Paixão); Hospital Egas Moniz, Lisbon (E. Gonçalves, C. Toscano); Narodowy Instytut Leków, Warsaw, Poland (J. Empel, M. Urbaś); Hospital Universitari de Bellvitge, Barcelona, Spain (M.A. Domínguez); University of Copenhagen, Copenhagen, Denmark (H. Westh); The Rockefeller University, New York, New York, USA (H. de Lencastre)
Main Article
Figure 5
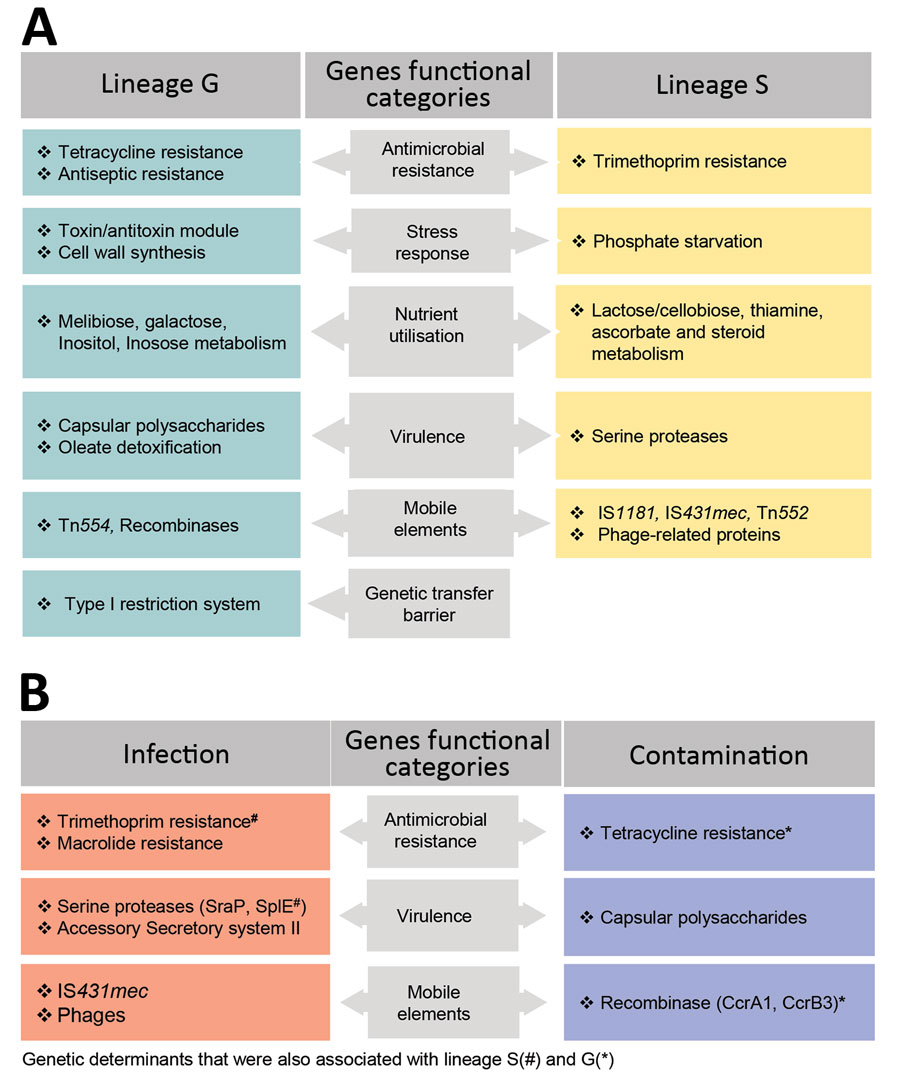
Figure 5. Genetic determinants that contribute to the distinction of clonal lineages and lifestyle of Staphylococcus saprophyticus. The graph displays determinants that contribute (A) and mediate (B) adaptation of S. saprophyticus to either infection or contamination. We used the genome-wide association study (GWAS) method to identify genetic factors by using 2 association comparisons: lineage G versus lineage S and human infection versus surface contamination. We used the pairwise comparison and included a core-SNP phylogenetic tree without recombination to remove the lineage effect in the analysis. Hits with Benjamini Hochberg corrected p<0.05 and odds ratio >1 were considered statistically significant. We grouped the identified genes into biologic functions based on gene annotation predicted by Prokka (https://vicbioinformatics.com/software.prokka.shtml). Some genetic factors that were associated with infections and contamination also were associated with the lineages despite subjecting the GWAS to lineage correction.
Main Article
Page created: December 29, 2020
Page updated: February 21, 2021
Page reviewed: February 21, 2021
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.