Volume 28, Number 1—January 2022
Research
Systematic Genomic and Clinical Analysis of Severe Acute Respiratory Syndrome Coronavirus 2 Reinfections and Recurrences Involving the Same Strain
Cite This Article
Citation for Media
Abstract
Estimates of the burden of severe acute respiratory syndrome coronavirus 2 reinfections are limited by the scarcity of population-level studies incorporating genomic support. We conducted a systematic study of reinfections in Madrid, Spain, supported by genomic viral analysis and host genetic analysis, to cleanse laboratory errors and to discriminate between reinfections and recurrences involving the same strain. Among the 41,195 cases diagnosed (March 2020–March 2021), 93 (0.23%) had 2 positive reverse transcription PCR tests (55–346 days apart). After eliminating cases with specimens not stored, of suboptimal sequence quality, or belonging to different persons, we obtained valid data from 22 cases. Of those, 4 (0.01%) cases were recurrences involving the same strain; case-patients were 39–93 years of age, and 3 were immunosuppressed. Eighteen (0.04%) cases were reinfections; patients were 19–84 years of age, and most had no relevant clinical history. The second episode was more severe in 8 cases.
Since the first description of a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reinfection on August 24, 2020, in a patient from Hong Kong (1) who acquired a second infection after having traveled to Europe, several reports have described other individual reinfection cases in different countries. These cases suggest the lack of a common reinfection pattern, with a variety of intervals between episodes, severity of episodes, clinical history, etc (2–5).
Genomic viral analysis has been applied to determine within-host SARS-CoV-2 evolution in patients with persistent infection (6,7) but has not been used in the same way to analyze coronavirus disease (COVID-19) recurrences involving the same strain. The scant available reports focus primarily on clinical descriptions (8,9), only some of which are supported by detailed genomic analysis (10).
Although a reasonable number of studies have analyzed individual COVID-19 recurrences in detail, population-level studies addressing this issue more systematically are lacking. We present a systematic analysis of all COVID-19 recurrences diagnosed at a tertiary hospital in Madrid, Spain (320,956 case-patients, 11.3% of the total Madrid population), over a 12-month period. Our analysis was supported by genomic viral analysis, cleansing of laboratory errors by host genetic analysis, consideration of both reinfections and recurrences involving the same strain, and integrating clinical features of the cases.
Patients and Methods
The study period was March 2020−March 2021. The cases selected for study were required to have 2 sequential positive reverse transcription PCR (RT-PCR) tests taken >45 days apart with >1 negative RT-PCR between positive tests. When the interepisode interval was >120 days and a different lineage was involved in each episode, the negative RT-PCR between episodes was not obligatory.
Specimens
The specimens corresponded to the remnants of nasopharyngeal swabs taken for diagnostic purposes. Specimens were stored at −70°C until analysis.
Clinical Data
The baseline characteristics, clinical and laboratory parameters at COVID-19 diagnosis, and outcomes of patients were obtained from their electronic medical records. The study was approved by the ethical research committee of Gregorio Marañón Hospital, Madrid (REF: MICRO.HGUGM.2020–042).
Diagnostic Tests
SARS-CoV-2 RT-PCRs and Serology
We extracted and purified viral RNA from 300 μL of nasopharyngeal exudates with the KingFisher instrument (ThermoFisher Scientific, https://www.thermofisher.com). This process was followed by RT-PCR using the TaqPath COVID-19 CE-IVD RT-PCR kit (ThermoFisher Scientific), which targets open reading frame 1ab, nucleoprotein, and spike genes. We performed serum antibody determinations by specific quantitative detection of SARS-CoV-2 IgG by using a chemiluminescent microparticle immunoassay on the ARCHITECT system (SARS-CoV-2 IgG II Quant Reagent Kit; Abbott Laboratories, https://www.abbott.com).
Whole-Genome Sequencing
We used 11 μL of RNA as template for reverse transcription using Invitrogen SuperScript IV reverse transcription and random hexamers (both ThermoFisher Scientific). We performed whole-genome amplification of the coronavirus with an Artic_nCov-2019_V3 panel of primers (Integrated DNA Technologies, Inc., https://www.idtdna.com) (https://artic.network/ncov-2019) and Q5 Hot Start DNA polymerase (New England Biolabs, https://www.neb.com). We prepared libraries by using the Nextera DNA Flex Library Preparation Kit (Illumina, https://www.illumina.com), following the manufacturer’s instructions.
Libraries were quantified with a Quantus Fluorometer (Promega, https://www.promega.com) before being pooled at equimolar concentrations (4 nM). We then sequenced them in pools of <17 libraries on the Miseq system (Illumina Inc.) with the MiSeq Reagent Micro kit version 2 (2×151 bp) or in pools of <96 libraries with the MiSeq Reagent (2×201 bp).
Sequences above the GISAID thresholds were deposited at GISAID (https://www.gisaid.org; Appendix 1 Table). An in-house analysis pipeline was applied on the sequencing reads (https://github.com/pedroscampoy/covid_multianalysis). In brief, the pipeline involves the following 4 steps: 1) removal of human reads with Kraken (11); 2) preprocessing and quality assessment of fastq files using fastp version 0.20.1 (12) (arguments: –cut tail, –cut-window-size, –cut-mean-quality, –max_len1, –max_len2) and fastQC version 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc); 3) mapping with BWA version 0.7.17 (13) and variant calling using IVAR v1.2.3 (14), using the Wuhan-1 SARS-CoV-2 sequence (GenBank accession no. NC_045512.2) as reference; and 4) calibration of occasional low coverage positions using joint variant calling. When necessary, we analyzed informative noncovered positions by standard Sanger sequencing by using the corresponding flanking primers from the ARTIC set.
Reinfections were considered when we detected a higher than expected number of single-nucleotide polymorphisms (SNPs) between the episodes (considering the standard estimation of 1 SNP/2 weeks) or a distribution of SNPs between the episodes consistent with independent evolutionary paths (SNPs present in the first episode but absent in the second episode and vice versa), or different variants or lineages involved in each episode or involvement in the second episode of a strain or variant that was not circulating in the population when the patient had the first episode. Recurrences were considered to involve the same strain when 0–1 SNPs were identified between the sequences from each episode.
Short Tandem Repeat Analysis
For human identity testing, we applied short tandem repeat (STR) PCR using the Mentype Chimera PCR amplification kit (Biotype, https://www.biotype.de) on the specimens used for SARS-CoV-2 genome sequencing. We examined 12 noncoding STR loci and the gender-specific amelogenin locus, labeled with 3 different dyes (6-FAM, BTG, or BTY). The selected loci had a very high rate of heterozygosity and balanced allelic distribution (15). We performed PCR with 0.2–1 ng of genomic DNA using the Mentype Chimera PCR amplification kit (Biotype), the GeneAmp PCR System 9700 Thermal Cycler, followed by capillary electrophoresis on a Genetic Analyzer 3130xl (both ThermoFisher Scientific), as recommended by the manufacturer.
Figure 1
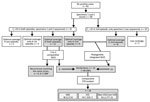
Figure 1. Flow diagram of the analysis and selection criteria for severe acute respiratory syndrome coronavirus 2 re-positive cases, Madrid, Spain, March 2020–March 2021. Re-positive cases were those that had 2 sequential...
The criteria for selecting SARS-CoV-2–positive cases for the study was 2 sequential positive RT-PCR tests taken >45 days apart with >1 negative RT-PCRs between the positive tests. Of the 41,195 cases diagnosed during the study period (March 2020–March 2021), 93 (0.23%) fulfilled these criteria, with positive specimens taken 55–346 days apart (Figure 1). We classified these cases as re-positive. Two specimens had been stored for each of 68 (73%) of the 93 re-positive cases, and of these, 32 (34%) were suitable for sequencing and comparison of sequences because cycle threshold (Ct) values for both positive specimens were <33 (Figure 1). The sequencing quality parameters of the 2 specimens were above the recommended threshold for a robust SNP calling (>90% of the genome with >30× coverage depth) in only 12 cases (29%). In another 17 cases, only 1 of the 2 specimens offered sequences of sufficient quality (Figure 1).
Recurrences Involving the Same Strain
After comparing the SNPs called in the sequences from the sequential episodes of the 12 re-positive cases, 4 (0.01% of total diagnosed cases) were classified as recurrences involving the same strain (Table 1) (0–1 SNPs between them; 3 belonged to A.5 lineage and 1 to Z.1 lineage [parental lineage: B.1.177.50]). Time between episodes ranged from 55 to 114 days, and Ct values for the second episode were consistent with active infection (Ct 19–28). All had 1 negative result between the positive SARS-CoV-2 RT-PCR test, and 1 also had a second intermediate negative test.
The 4 patients ranged from 39 to 93 years of age; underlying conditions were 1 heart transplant, 1 bone marrow transplant, 1 case of chronic renal insufficiency, and 1 case of obesity and high blood pressure (Table 1). Of the patients with underlying conditions, 3 had a clinical history of some degree of chronic immunosuppression: case-patient 1 underwent a heart transplant in June 2020 and was being treated with prednisone and mycophenolate, case-patient 3 had chronic kidney disease, and case-patient 4 underwent a bone marrow transplant in 2019 and was receiving treatment with sirolimus and ruxolitinib. Case-patient 2 had no known immunosuppression. Case-patients 1 and 4 seroconverted after the first SARS-CoV-2–positive episode (Table 1). Serologic testing was not available for case-patient 2, and case-patient 3 had a negative serologic result but was measured soon after the primary infection. For the second SARS-CoV-2 infection, case-patients 1, 2, and 4 seroconverted; results of serologic testing were not available for case-patient 3 (Table 1). In 2 cases, the second episode was milder in severity. In another case, both episodes were asymptomatic; for the remaining case-patient, who had a mild first episode, data were not available for the second infection. Two case-patients were asymptomatic between the 2 episodes, and the other 2 experienced asthenia and general malaise.
Reinfections
Figure 2
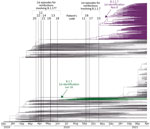
Figure 2. Global dating of the first emergence of severe acute respiratory syndrome coronavirus 2 variants identified in reinfections, Madrid, Spain, March 2020–March 2021, with available sequences only for the second specimen...
In 8 of the remaining re-positive cases, we identified 7–49 different SNPs between the sequences from the 2 sequential positive specimens, which indicated that they were reinfections (Table 2; Figure 1; Appendix 2 Figure). In addition to the standard approach to identifying reinfections (i.e., direct comparison of SNPs between SARS-CoV-2 sequences obtained in 2 sequential episodes), we also followed an alternative approach (16) using a population-based integrated phylogenetic approach to demonstrate that the second episode involved a strain that was not circulating in the population during the patient’s first episode. To apply this alternative strategy, we needed sequencing data only from the second episode of COVID-19. Therefore, we recovered the 9 cases from the second episode providing optimal sequence coverage that had been ruled out for 1-to-1 SNP comparisons (Figure 1). We were also able to add a further 8 cases with optimal sequences out of 10 cases with Ct values <33 in the second episode that had previously been ruled out for comparative sequencing (Figure 1). In 14 cases, we identified SARS-CoV-2 variants (9 B.1.177 and 5 B.1.1.7) with dates of emergence in our population after these patients experienced their first episodes (Figure 2). The first description in Spain for B.1.177 was June 16, 2020 (hCoV-19/Spain/IB-IBV-99010764/2020; GISAID accession no. EPI_ISL_691664) and for B.1.1.7 was November 8, 2020 (hCoV-19/Spain/VC-IBV-98012610/2020; accession no. EPI_ISL_1060510). This information indicates that the variants involved in these patients’ second episodes were not circulating in Spain at the time of their first episodes and therefore correspond to reinfections.
We subjected the 22 total reinfections assigned according to the standard or alternative phylogenetic approaches to a final validation to demonstrate that the specimens in the first and second episodes belonged to the same host and to rule out erroneous assignment of reinfections as a result of incorrect labeling or handling of samples. STR genetic analysis identified 2 pairs of specimens with genetic differences, whereas STR analysis of 2 specimens from 2 cases did not yield interpretable results; we eliminated all 4 cases from the study, leading to final validation of 18 reinfections (0.04% of total diagnosed cases and 81.82% of initially suspected reinfections by viral genomic analysis).
The positive specimens from the 18 reinfection cases validated by host genetic analysis were taken 116–342 days apart. Of these 18 cases, 6 reinfections involved the B.1.1.7 (Alpha) variant of concern, 1 involved the B.1.525 variant of interest, and the remaining 11 cases involved the B.1.177 variant (neither variant of concern nor variant of interest).
The age range for reinfected cases was 19 to 84 years of age. Most (13/18) had no relevant clinical antecedents (Table 3), and of those with underlying conditions, we highlight 1 renal transplant, 1 case-patient with asthma, 1 with chronic kidney disease, and 1 with autoimmune disease. In those for whom serologic data were available for the first and second episodes, SARS-CoV-2 serologic test results were positive in 2/9 first episode cases and 11/11 second episode cases (Table 3). For the first episode, 6 case-patients were asymptomatic, 6 had mild symptoms, 6 were moderately symptomatic, and no cases were severe. The second episode was mild in 11 cases, and only 1 case-patient was asymptomatic. Comparing the symptoms for the sequential episodes, the second episode was more severe in 8 cases (bilateral pneumonia occurred in 3 case-patients); symptoms were milder in 1 case and equivalent to the first episode in the remaining cases.
Since the first description of a SARS-CoV-2 reinfection (1), many reports have been published documenting single cases of reinfection (2–5) and demonstrating the wide variety of ages, clinical backgrounds, and severity among episodes (17). According to the European Centre for Disease Prevention and Control (ECDC), in the 12 European Union countries that reported cases, 1,887 likely reinfections in 2020 and 691 likely reinfections from January–February 2021 were under investigation (18).
Despite the large number of reports communicating SARS-CoV-2 reinfections, they are rare, although estimates of the true impact are limited by the scarcity of larger population-level studies. A nationwide study performed in Denmark (19) concluded that 0.65% of SARS-CoV-2–positive cases during the first COVID-19 wave had a second positive test in the second wave, and that this percentage increased to 3.27% in those with a negative result in the first wave. These data allowed Hansen et al. (19) to infer that protection against repeat infection in those who had natural immunity from previous SARS-CoV-2 infection was 80.5%, decreasing to 47.1% among persons >65 years of age.
Other studies have tried to go beyond the reporting of single cases by offering data on the frequency of SARS-CoV-2 reinfections in different countries; results range from 0.14 to 2.11% (19–24). However, in all these studies, the assignment of reinfections was supported only by sequential positive RT-PCR results, which means that, strictly speaking, these re-positive SARS-CoV-2 infections were considered suspected reinfections (22) without determining whether they were recurrences involving the same strain, reinfections, persistent cases, or testing errors (25). Assigning re-positive cases to 1 of the above categories is only possible when whole sequencing data are also included in the analysis.
The aim of our study was to overcome these limitations by enhancing the robustness of a systematic study of all COVID-19 cases diagnosed in our population, with the added value of a refined genomic analysis and considering both viral genomic analysis and host genetic analysis. This design makes it possible to precisely assign recurrences involving the same strain and reinfections and to cleanse test errors, in short of being able to offer solid data on the actual burden of these events in our population. Equivalent efforts should be made to study the impact of these events in other communities.
The percentage of re-positive cases we observed before genomic analysis (0.23%) is similar to that observed in other settings (26,27). To consider a case re-positive, we established a threshold of 45 days between 2 SARS-CoV-2–positive RT-PCR tests with >1 intermediate negative RT-PCR result, although in 69 of our 93 re-positive cases (74.2%), the episodes were >90 days apart.
Despite efforts to store specimens since the beginning of the pandemic, in 27% of the 93 re-positive cases, >1 of the 2 specimens were not available in our biobank, illustrating a main challenge of documenting reinfections (17). In addition to loss of cases, a second challenge was obtaining high-quality sequencing data, which was achieved in only one third of the cases with available specimens. In our experience processing recent specimens, the percentage of specimens with Ct values <33 that yielded suboptimal sequencing data was much lower (7%–10%). This experience serves as a cautionary warning of the potential deterioration of valuable remaining diagnostic specimens, even at −80° C, for future studies.
After comparative viral genomic analysis, identification of recurrences involving the same strain accounted for a reduction of 18.2%, and host genetic analysis a further 9.1% reduction (because specimens came from different persons), in the number of re-positive cases that would otherwise have been wrongly assigned as reinfections. On the basis of this finding, we also eliminated from the study another 2 cases with suboptimal results in the host genetic analysis, which did not enable us to draw conclusions. The dramatic increase in laboratory workload during the successive waves of COVID-19 infection likely led to mistakes in labeling samples or aliquoting. However, only a few studies that focused on documenting SARS-CoV-2 reinfections considered ruling out mislabeling of specimens by host genetic analysis (2,28). Our data indicate that a proportion of reinfections are more likely to be misassigned if genomic rigor is applied only to viral analysis and not to host analysis.
Of note, we used 2 approaches to assess reinfections. The first was the standard direct comparison of SARS-CoV-2 sequences, which revealed 6 reinfections, all but 1 differing by >20 SNPs (above the 2 SNPs/month estimated for SARS-CoV-2 evolution). The remaining reinfection differed by 7 SNPs, although the 7 differential SNPs were distributed in 3 SNPs that were specific to the first episode and not found in the second episode, and another 4 SNPs that were identified in the second episode but not in the first episode. This distribution of SNPs demonstrates that the second strain could not have evolved from the first one, consistent with reinfection. After the standard 1-to-1 comparative approach to identify reinfections, we applied a second alternative approach (16), based on a population-based integrated phylogenetic approach, to demonstrate that the strain involved in the reinfection had not yet emerged in our population at the time of the patient’s first episode. This alternative approach, in which we identified 12 additional reinfections, supports the need to expand the criteria for assigning SARS-CoV-2 reinfections, as the ECDC (29) did when it accepted the use of whole-genome sequencing to document reinfections by demonstrating that the strain involved in the reinfection was clustered with other strains circulating at the site of exposure (29). Considering the difficulties of storing all remaining specimens during the pandemic because of the high diagnostic workload, the alternative phylogenetic approach applied in this study could pave the way for more extensive documentation of the actual magnitude of reinfections in different populations.
A systematic review (25) concluded that reinfections were more likely to correspond to re-positive cases with a second positive RT-PCR >3 months after the first episode. Our reinfection data are consistent with this observation, because the time between episodes ranged from 116 to 346 days. Our data would fit the recent definition of a reinfection case by the ECDC (18), which establishes a 90-day threshold for reinfection to be considered.
The fact that most reported reinfections occurred >3 months after the first episode suggests the progressive decline in antibodies after a first infection plays a likely role. Unfortunately, in most studies, serologic data for first infections are lacking, which limits the documentation of this hypothesis. In our study, only 2 of 9 cases for which serologic data were available had positive SARS-CoV-2 serologic results, whereas all 11 seroconverted after the reinfection episode. Our data point to the lack of immune response mounted after the first infection being a more likely explanation for reinfection than a progressive decline in antibodies.
With respect to differences in severity between the first and second episodes in SARS-CoV-2 reinfections, situations vary widely (17). In our study, the second episode was generally more severe; we noted 6 asymptomatic, 6 mild, 6 moderate, and no severe first episodes versus 1 asymptomatic, 11 mild, 2 moderate, and 3 severe second episodes.
Not all re-positive cases >3 months after first infection should be assumed to correspond to reinfection. In our study, of the 4 recurrences identified that involved the same strain, 2 also occurred within this period, whereas the remaining 2 occurred 55 and 77 days after the first episode, beyond the threshold proposed as highly suggestive of nonreinfections (25,30).
SARS-CoV-2 recurrences involving the same strain have attracted much less attention than reinfections, possibly because of the lack of genomic resolution in most studies addressing reinfections with population-level values. Our data indicate that 18.2% of SARS-CoV-2 re-positive cases corresponded to recurrences involving the same strain, which would otherwise have been mislabeled as reinfections if genomic viral analysis had not been included. The second episode was equivalent or milder in terms of severity. Of recurrences involving the same strain, 3 corresponded to patients with some degree of immunosuppression (renal transplantation, bone marrow transplantation, and chronic kidney disease). The very few cases of SARS-CoV-2 recurrences involving the same strain reported in other studies supported by genomic analysis also occurred in immunosuppressed patients (D.A. Molina, unpub. data, https://www.researchsquare.com/article/rs-92286/v1; 8).
The robustness of our study’s systematic design was coupled with the value of its methodological refinement, which integrated genomic viral analysis and host genetic analysis. This design enabled us to cleanse data by eliminating laboratory errors and to offer precise data about the true burden and clinical features of SARS-CoV-2 reinfections and recurrences involving the same strain. We performed our analysis before the emergence of most SARS-CoV-2 variants of concern. Therefore, this study constitutes a valuable reference for forthcoming comparative studies addressing the burden of reinfections and recurrences involving the same strain in the context of new SARS-CoV-2 variants with immune escape potential.
Members of the Gregorio Marañón Microbiology-ID COVID-19 Study Group: Javier Adán-Jiménez, Luis Alcalá, Teresa Aldámiz, Roberto Alonso, Beatriz Álvarez, Ana Álvarez-Uría, Juan Berenguer, Elena Bermúdez, Emilio Bouza, Sergio Buenestado-Serrano, Almudena Burillo, Ana Candela, Raquel Carrillo, Pilar Catalán, Emilia Cercenado, Alejandro Cobos, Víctor Manuel de la Cueva, Cristina Díez, Jose Egido-Balzategui, Pilar Escribano, Agustín Estévez, Chiara Fanciulli, Alicia Galar, M. a Dolores García, Darío García de Viedma, Paloma Gijón, Adolfo González, Helmuth Guillén, Jesús Guinea, Laura Vanessa Haces, Marta Herranz, Martha Kestler, Juan Carlos López, Carmen Narcisa Losada, Marina Machado, Mercedes Marín, Pablo Martín, Javier Martín-Escolano, Andrea Molero-Salinas, Pedro Montilla, Patricia Muñoz, María Olmedo, Álvaro Otero-Sobrino, Belén Padilla, María Palomo, Francisco Parras, María Jesús Pérez-Granda, Laura Pérez-Lago, Leire Pérez, Sandra R. Maus, Elena Reigadas, Carla Margarita Rico-Luna, Cristina Rincón, Belén Rodríguez, Sara Rodríguez, Cristina Rodríguez-Grande, Adriana Rojas, María Jesús Ruiz-Serrano, Carlos Sánchez, Mar Sánchez, Julia Serrano, Pedro J Sola-Campoy, Francisco Tejerina, Maricela Valerio, M. Cristina Veintimilla, Lara Vesperinas, Teresa Vicente, Sofía de la Villa.
Ms. Rodríguez-Grande is a predoctoral researcher at the Instituto de Investigación Sanitaria Gregorio Marañón, Madrid, in the Clinical Microbiology and Infectious Diseases Service of the Gregorio Marañón Hospital. Her scientific interest is focused on molecular biology and genomic studies of pathogenic organisms.
Acknowledgments
We thank Janet Dawson for editing and proofreading assistance.
This work was supported by the Instituto de Salud Carlos III (Ref COV20/00140: SeqCOVID—Consorcio para la epidemiología genómica de SARS-CoV-2 en España) and by Consejo Superior de Investigaciones Científicas (CSIC) (PTI Salud Global). L.P.L. is the recipient of a Miguel Servet Research contract (CPII20/00001) from the Instituto de Salud Carlos III.
References
- To KK, Hung IF, Ip JD, Chu AW, Chan WM, Tam AR, et al. Coronavirus disease 2019 (COVID-19) re-infection by a phylogenetically distinct SARS-coronavirus-2 strain confirmed by whole genome sequencing. Clin Infect Dis. 2021;73:e2946–51. DOIPubMedGoogle Scholar
- Tillett RL, Sevinsky JR, Hartley PD, Kerwin H, Crawford N, Gorzalski A, et al. Genomic evidence for reinfection with SARS-CoV-2: a case study. Lancet Infect Dis. 2021;21:52–8. DOIPubMedGoogle Scholar
- Van Elslande J, Vermeersch P, Vandervoort K, Wawina-Bokalanga T, Vanmechelen B, Wollants E, et al. Symptomatic severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reinfection by a phylogenetically distinct strain. Clin Infect Dis. 2021;73:354–6. DOIPubMedGoogle Scholar
- Mulder M, van der Vegt DSJM, Oude Munnink BB, GeurtsvanKessel CH, van de Bovenkamp J, Sikkema RS, et al. Reinfection of severe acute respiratory syndrome coronavirus 2 in an immunocompromised patient: a case report. Clin Infect Dis. 2021;73:e2841–2. DOIPubMedGoogle Scholar
- Larson D, Brodniak SL, Voegtly LJ, Cer RZ, Glang LA, Malagon FJ, et al. A case of early re-infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2021;73:e2827–8. DOIPubMedGoogle Scholar
- Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N Engl J Med. 2020;383:2291–3. DOIPubMedGoogle Scholar
- Baang JH, Smith C, Mirabelli C, Valesano AL, Manthei DM, Bachman MA, et al. Prolonged severe acute respiratory syndrome coronavirus 2 replication in an immunocompromised patient. J Infect Dis. 2021;223:23–7. DOIPubMedGoogle Scholar
- Yadav SP, Thakkar D, Bhoyar RC, Jain A, Wadhwa T, Imran M, et al. Asymptomatic reactivation of SARS-CoV-2 in a child with neuroblastoma characterised by whole genome sequencing. IDCases. 2021;23:
e01018 . DOIPubMedGoogle Scholar - Coppola A, Annunziata A, Carannante N, Di Spirito V, Fiorentino G. Late reactivation of SARS-CoV-2: a case report. Front Med (Lausanne). 2020;7:531. DOIPubMedGoogle Scholar
- Pérez-Lago L, Aldámiz-Echevarría T, García-Martínez R, Pérez-Latorre L, Herranz M, Sola-Campoy PJ, et al.; On Behalf Of Gregorio Marañón Microbiology-Id Covid Study Group. Different within-host viral evolution dynamics in severely immunosuppressed cases with persistent SARS-CoV-2. Biomedicines. 2021;9:808. DOIPubMedGoogle Scholar
- Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. [</jrn>]. Genome Biol. 2014;15:R46. DOIPubMedGoogle Scholar
- Chens S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast-all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:1884–90.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. DOIPubMedGoogle Scholar
- Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8.
- Thiede C, Bornhäuser M, Ehninger G. Evaluation of STR informativity for chimerism testing—comparative analysis of 27 STR systems in 203 matched related donor recipient pairs. Leukemia. 2004;18:248–54. DOIPubMedGoogle Scholar
- Pérez Lago L, Pérez Latorre L, Herranz M, Tejerina F, Sola-Campoy PJ, Sicilia J, et al.; Gregorio Marañón Microbiology-ID COVID 19 Study Group. A complete analysis of the epidemiological scenario around a SARS-CoV-2 reinfection: previous infection events and subsequent transmission. MSphere. 2021;6:
e0059621 . DOIPubMedGoogle Scholar - Babiker A, Marvil CE, Waggoner JJ, Collins MH, Piantadosi A. The importance and challenges of identifying SARS-CoV-2 reinfections. J Clin Microbiol. 2021;59:e02769–20. DOIPubMedGoogle Scholar
- European Centre for Disease Prevention and Control. Reinfection with SARS-CoV-2: implementation of a surveillance case definition within the EU/EEA. 2021 [cited 2021 Apr 8]. https://www.ecdc.europa.eu/en/publications-data/reinfection-sars-cov-2-implementation-surveillance-case-definition-within-eueea
- Hansen CH, Michlmayr D, Gubbels SM, Mølbak K, Ethelberg S. Assessment of protection against reinfection with SARS-CoV-2 among 4 million PCR-tested individuals in Denmark in 2020: a population-level observational study. Lancet. 2021;397:1204–12. DOIPubMedGoogle Scholar
- Vitale J, Mumoli N, Clerici P, De Paschale M, Evangelista I, Cei M, et al. Assessment of SARS-CoV-2 reinfection 1 year after primary infection in a population in Lombardy, Italy. JAMA Intern Med. 2021;181:1407–8. DOIPubMedGoogle Scholar
- Graham MS, Sudre CH, May A, Antonelli M, Murray B, Varsavsky T, et al.; COVID-19 Genomics UK (COG-UK) Consortium. Changes in symptomatology, reinfection, and transmissibility associated with the SARS-CoV-2 variant B.1.1.7: an ecological study. Lancet Public Health. 2021;6:e335–45. DOIPubMedGoogle Scholar
- Slezak J, Bruxvoort K, Fischer H, Broder B, Ackerson B, Tartof S. Rate and severity of suspected SARS-Cov2 reinfection in a cohort of PCR-positive COVID-19 patients. Clin Microbiol Infect. 2021;
S1198-743X(21)00422-5 ; [Epub ahead of print].PubMedGoogle Scholar - Abu-Raddad LJ, Chemaitelly H, Malek JA, Ahmed AA, Mohamoud YA, Younuskunju S, et al. Assessment of the risk of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reinfection in an intense re-exposure setting. Clin Infect Dis. 2021;73:e1830–40. DOIPubMedGoogle Scholar
- Fabiánová K, Kynčl J, Vlčková I, Jiřincová H, Košťálová J, Liptáková M, et al. COVID-19 reinfections. Epidemiol Mikrobiol Imunol. 2021;70:62–7.PubMedGoogle Scholar
- Tang X, Musa SS, Zhao S, He D. Reinfection or reactivation of severe acute respiratory syndrome coronavirus 2: a systematic review. Front Public Health. 2021;9:
663045 . DOIPubMedGoogle Scholar - Abu-Raddad LJ, Chemaitelly H, Malek JA, Ahmed AA, Mohamoud YA, Younuskunju S, et al. Assessment of the risk of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reinfection in an intense reexposure setting. Clin Infect Dis. 2021;73:e1830–40. DOIPubMedGoogle Scholar
- Gautret P, Houhamdi L, Nguyen NN, Hoang VT, Giraud-Gatineau A, Raoult D. Does SARS-CoV-2 re-infection depend on virus variant? Clin Microbiol Infect. 2021;27:1374–5. DOIPubMedGoogle Scholar
- Lee JSKS, Kim SY, Kim TS, Hong KH, Ryoo NH, Lee J, et al. Evidence of severe acute respiratory syndrome coronavirus 2 reinfection after recovery from mild coronavirus disease 2019. Clin Infect Dis. 2021;73:e3002–8. DOIPubMedGoogle Scholar
- European Centre for Disease Prevention and Control ETAB. Threat assessment brief: reinfection with SARS-CoV-2: considerations for public health response. [cited 2020 Sep 21]. https://www.ecdc.europa.eu/en/publications-data/threat-assessment-brief-reinfection-sars-cov-2
- Tang X, Zhao S, He D, Yang L, Wang MH, Li Y, et al. Positive RT-PCR tests among discharged COVID-19 patients in Shenzhen, China. Infect Control Hosp Epidemiol. 2020;41:1110–2. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These senior authors contributed equally to this article.
2Members of the Gregorio Marañón Microbiology-ID COVID 19 Study Group are listed at the end of this article.
Table of Contents – Volume 28, Number 1—January 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Dario Garcia de Viedma, C/Dr. Esquerdo 46 (28007), Madrid, Spain
Top