Volume 28, Number 12—December 2022
Research
Emergence and Evolutionary Response of Vibrio cholerae to Novel Bacteriophage, Democratic Republic of the Congo1
Cite This Article
Citation for Media
Abstract
Cholera causes substantial illness and death in Africa. We analyzed 24 toxigenic Vibrio cholerae O1 strains isolated in 2015–2017 from patients in the Great Lakes region of the Democratic Republic of the Congo. Strains originating in southern Asia appeared to be part of the T10 introduction event in eastern Africa. We identified 2 main strain lineages, most recently a lineage corresponding to sequence type 515, a V. cholerae cluster previously reported in the Lake Kivu region. In 41% of fecal samples from cholera patients, we also identified a novel ICP1 (Bangladesh cholera phage 1) bacteriophage, genetically distinct from ICP1 isolates previously detected in Asia. Bacteriophage resistance occurred in distinct clades along both internal and external branches of the cholera phylogeny. This bacteriophage appears to have served as a major driver for cholera evolution and spread, and its appearance highlights the complex evolutionary dynamic that occurs between predatory phage and bacterial host.
Cholera remains an ongoing public health threat on the continent of Africa, especially in the Democratic Republic of the Congo (DRC), which in 2020 reported the largest number of cases (19,789) of any country in the world with the exception of Yemen (1). Biotype El Tor strains of the seventh cholera pandemic (P7ET) were first reported in Africa in the early 1970s (2–6). Major outbreaks occurred in DRC in 2008, 2009, 2011–2012, 2013, and 2015–2017; in 2017 alone, an estimated 53,000 cholera cases with 1,145 deaths were reported from 20 of 26 provinces in DRC (2). Outbreaks have been most persistent in the eastern part of DRC in the Great Lakes region, along the Albertine Rift (2–4).
As reported elsewhere (3), strains appear to have been initially introduced into this area as part of what has been characterized as the T5 introduction (1970–1972) under the first wave of P7ET. In 1992, as part of the third wave of P7ET, the disease was reintroduced by a strain from southern Asia, in what has been designated as the T10 introduction event (3). Subsequent studies have documented persistence of T10 strains in this region, and ongoing cholera outbreaks in the Great Lakes region and spread of strains from this area suggest establishment of a regional focus of endemic disease derived from the T10 introduction (6).
Bacteriophages (phages) and their host bacteria follow predator–prey dynamics that drive co-evolution, resulting in the long-term persistence of both within ecosystems (7). Phage predation has been linked with seasonal patterns of cholera emergence and with clinical response to infection in humans (8–11). Phages are generally highly specific to their host; thus, both phage and susceptible host must maintain a dynamic equilibrium to coexist. Recently, it has been shown that mobile genetic elements associated with sulfamethoxazole/trimethoprim (SXT) antimicrobial resistance, designated as SXT integrative conjugative elements (ICEs), can determine phage resistance in V. cholerae (12). Furthermore, another study has demonstrated that susceptibility to phage killing of marine V. lentus was mediated by as many as 6–12 mobile genetic elements (13). Taken together, these recent studies support the concept that phage/host in situ interplay has a major role in adaptation and evolution.
Using microbiologic, phylogenomic, and molecular clock analyses, we investigated endemic cholera in the DRC Great Lakes regional hotspot. We also explored the genetic resistance of these V. cholerae strains to a novel ICP1 (Bangladesh cholera phage 1) V. cholerae phage isolated in cholera patients in the region and genetically distinct from previous ICP1 phages detected in Asia (14,15).
Isolation and Characterization of Toxigenic V. cholerae O1 and Virulent Phages
In an initial study involving the isolation and characterization of toxigenic V. cholerae O1 strains, we collected fecal samples from suspected cholera patients admitted to cholera treatment centers around Goma, DRC, during 2015–2017 (Table). After collection, we brought the samples to the Laboratoire Provincial de Sante Publique du Nord-Kivu in Goma for microbiological and serologic analysis. We isolated bacteria and confirmed species using methods described elsewhere (16), then stored strains in soft Luria-Bertani (LB) Miller agar (0.7% agar) and sent them to the Emerging Pathogens Institute at the University of Florida (Gainesville, FL, USA) for sequencing.
In a second study, we tried to isolate phages preying on V. cholerae O1 strains from fecal samples obtained in 2016–2017 from 41 additional cholera patients. We centrifuged cholera rice-water fecal samples at 5,000 × g for 10 minutes and filtered resultant supernatant through a 0.22-μm syringe filter, stored them at 4°C in a sterile microfuge tube, and sent them to the Emerging Pathogens Institute for analysis. To identify virulent phages, we tested each filtered fecal sample using standard plaque assay against V. cholerae O1 AGC-15, a strain we randomly selected from the DRC isolates from the first part of the study (Table). AGC-15 has the wild-type ompU sequence, which encodes the receptor for ICP2; it also has the wild-type O1-antigen biosynthetic genetic region that serves as the receptor for ICP1 and ICP3 (14) and lacks any PLE elements mediating immunity to ICP1 (17). For phage purification, we picked a single clear plaque using a Pasteur pipette into 1 mL of LB broth and incubated it overnight at 4°C to enable the phage to diffuse out of the soft agar. We made high-titer stocks of purified phage by infecting AGC-15 with phage in LB broth culture.
Whole-Genome Mapping and High-Quality Single-Nucleotide Polymorphism Calling
We performed whole-genome sequencing on the 24 V. cholerae O1 isolates from the first part of the study with the Illumina MiSeq for 500 cycles (Qui); we further conducted high-quality single-nucleotide polymorphism (hqSNP) calling (Appendix). The final genomewide hqSNP alignment included 120 T10 sublineage V. cholerae genome sequences: 24 strains collected as part of our study (Table); 71 from publicly available genomes from outbreaks in eastern DRC during 2014–2016 (6); 6 archival and publicly available DRC genomes collected during 2001–2013; 17 genomes collected across Africa during 1998–2014; and 2 publicly available genomes from India, ancestors of T10 sublineage (3) (Appendix Table 1). We performed multilocus sequence typing analysis using the online tool PubMLST (K. Jolley, unpub data, https://doi.org/10.12688/wellcomeopenres.14826.1) (Appendix Table 2).
Phylogeography
All datasets used in this study passed phylogenetic quality checks (Appendix Figure 1). To explore the origins of strains in the eastern portion of DRC and neighboring countries we used the Bayesian phylogeographic coalescent-based method implemented in BEAST version 1.10.4 software (18–20). The reconstruction of V. cholerae O1 spatiotemporal spread from different locations through Bayesian phylogeography requires calibration of a molecular clock. We estimated evolutionary rates implementing a Hasegawa-Kishino-Yano nucleotide substitution model (21) with empirical base frequencies, gamma distribution of site-specific rate heterogeneity, and ascertainment bias correction (22), testing a constant demographic prior against nonparametric demographic models, Gaussian Markov random field Skyride (23) and Bayesian Skyline plot (24), to rule out spurious changes in effective population size inferred by a nonparametric model, which would, in turn, effect timing of divergence events (25). We obtained the weighted average of synonymous (dS) and nonsynonymous substitution rates (dN) in the protein-coding regions of the V. cholerae O1 genome for all internal and external branches from a subset of 200 Bayesian maximum credibility clade (MCC) trees randomly obtained from the posterior distribution of trees, as described elsewhere (26,27).
Whole genome sequencing, genome assembly and annotation of DRC phages
We sequenced 8 plaque-purified phages isolated from 8 independent patient fecal samples with Illumina MiSeq for 50 cycles. We obtained >200-fold coverage that helped with de novo assembly of each phage genome into 1 complete contig using CLC Genomics Workbench (QIAGEN, https://www.qiagen.com). We manually confirmed and corrected low-coverage or problem areas as needed to ensure authentic genome assembly. We annotated phage genomes as described elsewhere (15) and deposited sequences into the National Center for Biotechnology Information Sequence Read Archive (BioProject identification no. PRJNA748018; Appendix Table 3).
Figure 1
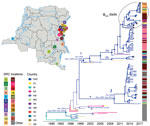
Figure 1. Spatiotemporal evolution and dissemination of Vibrio cholerae epidemic in the Democratic Republic of the Congo, 2015–2017. Sampling locations of V. choleraestrains sequenced in this study are...
Figure 2

Figure 2. Bayesian inference of the phylogenetic relationship between phages and mutation patterns in the Democratic Republic of the Congo and ICP1 (Bangladesh cholera phage 1) patterns from Asia. Sampling locations of...
Of the 24 toxigenic V. cholerae O1 strains isolated from fecal samples from cholera patients attending cholera treatment centers during 2015–2017 in the Goma region, 21 (87.5%) were serotype Inaba and 3 (12.5%) serotype Ogawa (Table). All strains in wave 3 were ctxB genotype-I and within the T10 introductory clade (3,6). Consistent with findings published elsewhere (3), our MCC tree (Figure 1; Appendix Figure 2) indicated a mean time for the most recent common ancestor (tMRCA) of the T10 V. cholerae sublineage introduced to Africa of March 1994 (95% highest posterior density [HPD] September 1991–February 1996). In addition, our analysis showed that subsequent independent introductions (spillover events denoted by asterisks in Figure 1) in the DRC Great Lakes region likely occurred from Rwanda. The first spillover, in May 2001 (95% HPD September 1999–June 2001), is represented by a DRC isolate, ERR1878097_CD_2003, that branches out of a lineage circulating in Rwanda (Figure 1). The other event resulted in 2 major monophyletic clades that match multilocus sequence types reported elsewhere (6).
The tMRCA of the first major lineage (denoted as I in Figure 1), February 2009 (95% HPD November 2005–September 2011), corresponds to the sequence type 69 cluster (6) identified during the first reported outbreaks of cholera in DRC during 2008 and 2009. This cluster included the only Ogawa serotype isolates present in our collection. However, the long branch separating the isolate from the Tanganyika province (ERR572559_CD_2013) strain at the base of the monophyletic clade raises the possibility of unsampled V. cholerae strains (either from Rwanda or other neighboring countries) that could constitute missing linkage between this DRC lineage and its actual ancestor. The second major lineage (denoted as II in Figure 1) contains all of the Inaba serotype strains collected. According to the molecular clock calibration, lineage II tMRCA dates to September 1999 (95% HPD March 1999–September 2000). The monophyletic clade also includes 3 strains identified in Zambia and Niger, which were likely the result of spillover events from DRC. This clade further divides into 2 sublineages (IIa and IIb in Figure 1) that diverged in November 2004 (95% HPD September 2002–March 2007). Overall, molecular clock and phylogeographic reconstruction suggests circulation of cholera lineages in DRC years before the first reported cholera outbreaks in 2008–2009.
Phages were isolated from 17/41 (41.5%) fecal samples screened, on the basis of formation of plaques on strain AGC_15_CD_2017. Whole-genome sequencing of a subset (n = 8) of these phages showed that they shared high similarity in sequence (hqSNPs = 114) and diverged substantially (hqSNPs = 8,441) from the ICP1 phage isolated from Bangladesh and India (Figure 2). Previously, a total of 185 core open reading frames were identified as being conserved in ICP1 isolates collected over a 12-year period in Bangladesh and India (15). Uniquely, the DRC ICP1 lacks 15 of these core open reading frames and also has 10.6 kb of novel sequence in the first third of the genome. Of particular note, genomes do have the anti-Vchind5 factor OrbA (12). However, like most ICP1-encoded gene products, most genes unique to DRC ICP1 are classified as hypothetical proteins because of a lack of an informative BLAST identification.
We screened our 24 DRC V. cholerae O1 strains for susceptibility to DRC ICP1 by plaque assays using ICP1_2017_A_DRC as a reference phage. Eighteen (75%) of the 24 V. cholerae strains were susceptible to ICP1_2017_A_DRC (Table); we observed that 2 strains had turbid plaques on plaque assay, as described elsewhere (28). At the genome level, all resistant strains, and 3 of 6 sensitive strains, including the 2 strains that produced turbid plaques, carried >1 mutation in genes that belong to the O1-antigen biosynthetic gene cluster. ICP1 uses the O1 antigen as its receptor, and V. cholerae is known to undergo phase variation to decrease or produce modified forms of the O1 antigen to evade ICP1 infection (7). However, the fact that resistant strains gave a positive serologic response when tested for the O1 antigen suggests that there are mechanisms for resistance to ICP1 that lie elsewhere in the genome.
SXT-ICE has been reported to have 5 hotspots within the accessory region, including hotspot 5 (VchInd5), which confers resistance to ICP1 phage infection (12). When we evaluated the SXT-ICE sequence in all 24 DRC V. cholerae genomes, we found that all harbored genes identical to the wild-type SXT-ICE, which should make the strains phage-resistant. However, as already noted, the DRC ICP1 phage that we identified encodes the anti-BREX factor OrbA, which protects against the host VchInd5 (12). We did not detect other mechanisms usually associated with resistance of host cells to phage, such as acquisition and expression of a family of phage-inducible chromosomal island–like elements (17), and none of the DRC ICP1 isolates encoded a previously described CRISPR-cas system specifically targeting phage-inducible chromosomal island–like elements for destruction, which might enable the phage to evade host immunity (29). At this point we cannot comment further on the mechanisms underlying resistance of our DRC V. cholerae strains to the regional DRC ICP1 phage other than to note the complexity of these regional phage/host interactions.
Figure 3
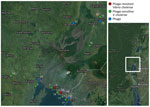
Figure 3. Sampling locations of phages and phage-resistant or sensitive Vibrio choleraeisolates in the Democratic Republic of the Congo. Each sampling location is coded by color (key) and number, which...
Comparison of genome-wide weighted averages of dS and dN along the internal branches of the cholera phylogeny showed a dN/dS ratio significantly >1 (p<0.001) (Appendix Figure 3, panel A). Moreover, the difference between dN and dS divergence accumulating over time along the internal branches of the phylogeny also appears to be increasing (Appendix Figure 3, panel B). In other words, the mixed presence of susceptible and resistant V. cholerae phenotypes, at least in the DRC Goma region where samples were collected, together with dN/dS patterns suggest that V. cholerae has been evolving under pressure of increasing diversifying selection, possibly driven by the co-circulation of predatory phages. Indeed, the map of sampling locations shows that phage-resistant or phage-susceptible V. cholerae strains, as well as independently sampled phages, have tended to co-circulate in the DRC Goma region and surrounding locales (Figure 3).
Figure 4

Figure 4. Phage-resistant or phase-sensitive dynamics and mutational patterns of Vibrio choleraeisolates in the Democratic Republic of the Congo. The boxes show the mutations that have been found along the...
To examine in more detail the adaptive fitness landscape that might confer either resistance or sensitivity to phage predation, we optimized an MCC tree for the subset of V. cholerae sequences including all strains in the ϕR/S clade, as well as 2 outgroup strains, AGC-2-CD-2015 and AGC-8-CD-2015, that clustered outside the clade (Figure 4). We used a Bayesian phylogeographic model with phage resistance or susceptibility as discrete phenotypic characters to infer the most likely phenotype of the ancestral (internal) nodes of the tree. The analysis clearly shows that the backbone path (trunk) of the ϕR/S clade, which represents the surviving lineage successfully propagating through time (28), is dominated by isolates with the phage-sensitive phenotype and connects phage-sensitive ancestral sequences that first generated a subcluster of strains circulating in 2015–2016 and then a subcluster including 2017 strains.
We cannot say which mutations are responsible for acquisition of phage resistance, but mutations in genes belonging to the O1-antigen biosynthetic gene cluster appear to have emerged, independently, along 3 distinct evolutionary lineages. The first lineage, leading to strain AGC-6-2015-DRC sampled in 2015, is characterized by amino acid substitutions in the rfbV, rpIE, phrA, and fliD genes. The second lineage resulted in a monophyletic clade of phage-resistant strains with mutations in either rfbN (strains sampled in 2015) or rfbD (sampled in 2016). The third lineage, leading to strain AGC-23-2017-DRC (sampled in 2017), was characterized again by an amino acid substitution in the rfbB gene.
Cholera continues to be a major public health problem in the Great Lakes region of Africa (1–4). To optimize cholera control and appropriately target public health interventions, evolutionary drivers for V. cholerae in this area need to be determined, including reasons why certain V. cholerae strains emerge and persist while others fail to propagate. Our data provide further information on sources and subsequent development of endemic V. cholerae O1 in eastern DRC. Our work also highlights the effects a novel regional bacteriophage can have on cholera evolution. As reflected in the trunk of the phylogeny of the ϕR/S clade, V. cholerae isolates displaying the phage-sensitive phenotype appear to be successfully propagating, with every branch that leads to phage-resistant phenotypes in the phylogeny eventually dying out. Our findings are somewhat counterintuitive: phage resistance, rather than encouraging expansion of the epidemic clone, led to evolutionary dead ends; however, our data highlight the ability of V. cholerae to explore and quickly abandon different evolutionary pathways during epidemic spread. This finding is not surprising considering the potential fitness cost of phage resistance, particularly if resistance results in mutants highly attenuated for virulence (30,31). Further work will be needed to determine the exact mechanism by which the V. cholerae strains isolated in this study were either completely or partially resistant (turbid plaque) to ICP1_2017_A_DRC and whether the phage can mutate to regain virulence (12).
In summary, our study documents a complex co-evolutionary dynamic involving V. cholerae and predatory phages (30) in the Great Lakes region of DRC. Phage-sensitive and highly infectious strains co-circulate with phage-resistant ones that occasionally emerge and eventually die out along different evolutionary pathways in response to the presence or absence of predatory phages in the environment, although the main phage-sensitive evolutionary lineage continued to propagate over time. The ability of V. cholerae to explore multiple mutational pathways in different genes and achieve phage resistance provides a substantial evolutionary advantage in terms of quick adaptive response to a changing environment, leading to emergence of new strains. Continuous monitoring of toxigenic V. cholerae and predator ICP1 phages in both patient fecal samples and aquatic environments in DRC and elsewhere could provide invaluable epidemiologic data for monitoring the spread of cholera, identifying environmental actors driving successful dissemination, and assessing the potential for new outbreaks.
Dr. Alam is a biological scientist in the University of Florida College of Public Health and Health Professions and the Emerging Pathogens Institute. His research focuses on the factors and processes promoting persistence of Vibrio cholerae in aquatic reservoirs and evolution resulting from the lytic bacteriophage–V. cholerae in both clinical and environmental settings. Dr. Mavian is on the faculty at the University of Florida College of Medicine in the Department of Pathology, Immunology, and Laboratory Medicine. Her research focuses on intrahost evolution of HIV, population dynamics of cholera, and evolution and global spread of arboviruses and, since the beginning of the pandemic, the dynamics of SARS-CoV-2.
Acknowledgments
R scripts and XML files are available from the authors upon request.
This work was supported by a National Institutes of Health/National Institute of Allergies and Infectious Disease–sponsored grant (R01 AI138554 to J.G.M., Jr.)
A. Ali, A.C., C.M., J.G.M., and M.S. conceived and designed the experiments; A. Angermeyer, A.M., C.M., K.K.S., M.N.C., and T.K.P. performed the experiments; A.M., C.M., F.M.M., K.D.S., M.S.T., and T.K.P. analyzed the data; A. Ali, A.C., F.M.M., J.G.M., and R.K.K.S. contributed materials/analysis tools; A. Ali, A.C., C.M., J.G.M., and M.S. wrote the paper.
References
- World Health Organization. Cholera annual report 2020. Wkly Epidemiol Rec. 2021;96:445–60.
- Ingelbeen B, Hendrickx D, Miwanda B, van der Sande MAB, Mossoko M, Vochten H, et al. Recurrent cholera outbreaks, Democratic Republic of the Congo, 2008–2017. Emerg Infect Dis. 2019;25:856–64. DOIPubMedGoogle Scholar
- Weill FX, Domman D, Njamkepo E, Tarr C, Rauzier J, Fawal N, et al. Genomic history of the seventh pandemic of cholera in Africa. Science. 2017;358:785–9. DOIPubMedGoogle Scholar
- Okeke IN. Africa in the time of cholera: a history of pandemics from 1817 to the present [book review]. Emerg Infect Dis. 2012;18:362. DOIGoogle Scholar
- Moore S, Miwanda B, Sadji AY, Thefenne H, Jeddi F, Rebaudet S, et al. Relationship between distinct African cholera epidemics revealed via MLVA haplotyping of 337 Vibrio cholerae isolates. PLoS Negl Trop Dis. 2015;9:e0003817–0003817. DOIPubMedGoogle Scholar
- Irenge LM, Ambroise J, Mitangala PN, Bearzatto B, Kabangwa RKS, Durant JF, et al. Genomic analysis of pathogenic isolates of Vibrio cholerae from eastern Democratic Republic of the Congo (2014-2017). PLoS Negl Trop Dis. 2020;14:
e0007642 . DOIPubMedGoogle Scholar - Seed KD. Battling phages: how bacteria defend against viral attack. PLoS Pathog. 2015;11:
e1004847 . DOIPubMedGoogle Scholar - Faruque SM, Naser IB, Islam MJ, Faruque AS, Ghosh AN, Nair GB, et al. Seasonal epidemics of cholera inversely correlate with the prevalence of environmental cholera phages. Proc Natl Acad Sci U S A. 2005;102:1702–7. DOIPubMedGoogle Scholar
- Huq A, Sack RB, Nizam A, Longini IM, Nair GB, Ali A, et al. Critical factors influencing the occurrence of Vibrio cholerae in the environment of Bangladesh. Appl Environ Microbiol. 2005;71:4645–54. DOIPubMedGoogle Scholar
- Silva-Valenzuela CA, Camilli A. Niche adaptation limits bacteriophage predation of Vibrio cholerae in a nutrient-poor aquatic environment. Proc Natl Acad Sci U S A. 2019;116:1627–32. DOIPubMedGoogle Scholar
- Seed KD, Yen M, Shapiro BJ, Hilaire IJ, Charles RC, Teng JE, et al. Evolutionary consequences of intra-patient phage predation on microbial populations. eLife. 2014;3:
e03497 . DOIPubMedGoogle Scholar - LeGault KN, Hays SG, Angermeyer A, McKitterick AC, Johura FT, Sultana M, et al. Temporal shifts in antibiotic resistance elements govern phage-pathogen conflicts. Science. 2021;373:
eabg2166 . DOIPubMedGoogle Scholar - Hussain FA, Dubert J, Elsherbini J, Murphy M, VanInsberghe D, Arevalo P, et al. Rapid evolutionary turnover of mobile genetic elements drives bacterial resistance to phages. Science. 2021;374:488–92. DOIPubMedGoogle Scholar
- Seed KD, Bodi KL, Kropinski AM, Ackermann HW, Calderwood SB, Qadri F, et al. Evidence of a dominant lineage of Vibrio cholerae-specific lytic bacteriophages shed by cholera patients over a 10-year period in Dhaka, Bangladesh. MBio. 2011;2:e00334–10. DOIPubMedGoogle Scholar
- Angermeyer A, Das MM, Singh DV, Seed KD. Analysis of 19 highly conserved Vibrio cholerae bacteriophages isolated from environmental and patient sources over a twelve-year period. Viruses. 2018;10:10. DOIPubMedGoogle Scholar
- Ali A, Chen Y, Johnson JA, Redden E, Mayette Y, Rashid MH, et al. Recent clonal origin of cholera in Haiti. Emerg Infect Dis. 2011;17:699–701. DOIPubMedGoogle Scholar
- O’Hara BJ, Barth ZK, McKitterick AC, Seed KD. A highly specific phage defense system is a conserved feature of the Vibrio cholerae mobilome. PLoS Genet. 2017;13:
e1006838 . DOIPubMedGoogle Scholar - Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLOS Comput Biol. 2009;5:
e1000520 . DOIPubMedGoogle Scholar - Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–32. DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74. DOIPubMedGoogle Scholar
- Leaché AD, Banbury BL, Felsenstein J, de Oca AN, Stamatakis A. Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst Biol. 2015;64:1032–47. DOIPubMedGoogle Scholar
- Minin VN, Bloomquist EW, Suchard MA. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol. 2008;25:1459–71. DOIPubMedGoogle Scholar
- Strimmer K, Pybus OG. Exploring the demographic history of DNA sequences using the generalized skyline plot. Mol Biol Evol. 2001;18:2298–305. DOIPubMedGoogle Scholar
- Hall MD, Woolhouse ME, Rambaut A. The effects of sampling strategy on the quality of reconstruction of viral population dynamics using Bayesian skyline family coalescent methods: A simulation study. Virus Evol. 2016;2:
vew003 . DOIPubMedGoogle Scholar - Lemey P, Kosakovsky Pond SL, Drummond AJ, Pybus OG, Shapiro B, Barroso H, et al. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLOS Comput Biol. 2007;3:
e29 . DOIPubMedGoogle Scholar - Mavian C, Paisie TK, Alam MT, Browne C, Beau De Rochars VM, Nembrini S, et al. Toxigenic Vibrio cholerae evolution and establishment of reservoirs in aquatic ecosystems. Proc Natl Acad Sci U S A. 2020;117:7897–904. DOIPubMedGoogle Scholar
- Seed KD, Faruque SM, Mekalanos JJ, Calderwood SB, Qadri F, Camilli A. Phase variable O antigen biosynthetic genes control expression of the major protective antigen and bacteriophage receptor in Vibrio cholerae O1. PLoS Pathog. 2012;8:
e1002917 . DOIPubMedGoogle Scholar - Seed KD, Lazinski DW, Calderwood SB, Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489–91. DOIPubMedGoogle Scholar
- Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog. 2013;9:
e1003800 . DOIPubMedGoogle Scholar - Oechslin F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses. 2018;10:10. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: November 10, 2022
1Previously presented at Epidemics—8th International Conference on Infectious Diseases Dynamics [online], November 30–December 3, 2021.
2These authors contributed equally to this article.
Table of Contents – Volume 28, Number 12—December 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Afsar Ali and Carla Mavian, Emerging Pathogens Institute, 2055 Mowry Rd, University of Florida, Gainesville, FL 32601, USA
Top