Volume 31, Number 1—January 2025
Research
Trichuriasis in Human Patients from Côte d’Ivoire Caused by Novel Species Trichuris incognita with Low Sensitivity to Albendazole/Ivermectin Combination Treatment
Cite This Article
Citation for Media
Abstract
Albendazole/ivermectin combination therapy is a promising alternative to benzimidazole monotherapy alone for Trichuris trichiura control. We used fecal DNA metabarcoding to genetically characterize Trichuris spp. populations in patient samples from Côte d’Ivoire showing lower (egg reduction rate <70%) albendazole/ivermectin sensitivity than those from Laos and Tanzania (egg reduction rates >98%). Internal transcribed spacer (ITS) 1 and ITS2 metabarcoding revealed the entire detected Côte d'Ivoire Trichuris population was phylogenetically distinct from T. trichiura found in Laos and Tanzania and was more closely related to T. suis. Mitochondrial genome sequencing of 8 adult Trichuris worms from Côte d’Ivoire confirmed their species-level differentiation. Sequences from human patients in Cameroon and Uganda and 3 captive nonhuman primates suggest this novel species, T. incognita, is distributed beyond Côte d'Ivoire and has zoonotic potential. Continued surveillance by using fecal DNA metabarcoding will be needed to determine Trichuris spp. geographic distribution and control strategies.
Trichuris trichiura is a soil-transmitted helminth infecting 465 million persons globally, primarily in middle and low-income countries (1). Infections are most prevalent in children; moderate to severe infections cause chronic dysentery, diarrhea, and stunted growth (2). Control of this parasitic infection is largely dependent on preventive chemotherapy in high-risk disease-endemic regions by administering albendazole and mebendazole annually or biannually (3,4). Benzimidazole efficacy against T. trichiura is generally low; a meta-analysis comparing 38 clinical trials reported T. trichiura egg reduction rates (ERRs) were ≈50% for albendazole and ≈66% for mebendazole, and cure rates were ≈30% for albendazole and ≈42% for mebendazole (5). However, albendazole/ivermectin combination therapy improved efficacy against T. trichiura (6–11). Consequently, the World Health Organization has added this drug combination to its Essential Medicines List for soil-transmitted helminths (3,11).
A double-blind, parallel-group, phase 3, randomized controlled clinical trial recently showed an expected high efficacy of albendazole/ivermectin combination therapy against T. trichiura in Laos (ERR 99%), and Pemba Island, Tanzania (ERR 98%). However, albendazole/ivermectin combination therapy showed an unexpectedly low efficacy in Cȏte d’Ivoire (ERR 70%) (12).
Community-scale genetic analysis of soil-transmitted helminths is challenging because harvesting large numbers of parasites from patients is labor intensive and can be logistically difficult, whether harvesting adult worms from expulsion studies or parasite eggs from feces. DNA metabarcoding is a technique that enables detection of multiple taxa or genotypes from a single environmental sample through high-throughput DNA sequencing; applied directly to fecal DNA, this method offers a powerful alternative approach for helminth analysis. However, eggs from worms belonging to the genus Trichuris are robust and difficult to disrupt within fecal matter; together with the presence of fecal PCR inhibitors, egg processing is a challenge for PCR and metabarcoding methods applied directly to fecal DNA (13). Multiple studies have aimed to improve the amplification of Trichuris DNA from human fecal samples by including a bead-beating step to mechanically disrupt the eggs (14–17). We used a DNA extraction protocol that combined multiple freeze–thaw cycles and mechanical disruption and applied metabarcoding to internal transcribed spacer (ITS) 1 and ITS2 rRNA gene regions to genetically characterize Trichuris populations in fecal samples.
DNA Extraction
We examined fecal samples preserved in ethanol from patients enrolled in a previously reported clinical trial (12). We prepared DNA from pretreatment fecal samples collected from patients in Cȏte d’Ivoire (n = 22), Laos (n = 36), and Pemba Island, Tanzania (n = 29). Trichuris egg counts were 91–1,151 eggs per gram (EPG) of feces (Appendix Table 1). We washed ≈250 mg of each fecal sample (in 90% ethanol) with molecular-grade water 3 times (1:2 ratio of feces:water) and centrifuged at 12,000 × g. We then performed 3 cycles of snap freezing in liquid nitrogen and 15 minutes of heating at 100°C with shaking at 750 rpm followed by vigorous bead beating for 3 minutes by using a Mini-Beadbeater-96 (Thomas Scientific, https://www.thomassci.com). We extracted DNA by using the QIAamp PowerFecal Pro DNA Kit (QIAGEN, https://www.qiagen.com) (Appendix).
We picked whole worms directly from patients’ fecal samples after anthelmintic treatment during an expulsion study in Côte d’Ivoire (M.A. Bär et al., unpub. data, https://doi.org/10.1101/2024.06.11.598441). We washed worms with sterile phosphate-buffered saline and stored them in absolute ethanol. We extracted DNA from separated worm heads by using a DNeasy Blood and Tissue Kit (QIAGEN).
Fecal DNA Metabarcoding
We designed PCR primers to amplify the following Trichuris genetic markers in fecal DNA: ITS1 (733-bp amplicon), ITS2 (592 bp), cox-1 (430 bp), nad-1 (470 bp), and nad-4 (446 bp) (Appendix Table 2). Each PCR reaction comprised 5 μL of 5X KAPA HiFi buffer (Roche, https://www.roche.com), 0.75 μL of 10 μmol/L Illumina-adapted forward primer and 0.75 μL of 10 μmol/L Illumina-adapted reverse primer (Illumina, https://www.illumina.com), 0.75 μL of 10 μmol/L dNTPs (Roche), 0.5 μL KAPA HiFi Hotstart polymerase (0.5 units) (Roche), 0.1 μL bovine serum albumin (Thermo Fisher Scientific, https://www.thermofisher.com), 15.15 μL molecular-grade water, and 2 μL of extracted fecal DNA. Thermocycling conditions were 95°C for 3 minutes, followed by 40 cycles of 98°C for 20 seconds, 65°C (ITS-1), 64°C (ITS-2, nad-1, nad-4), 60°C (cox-1), or 63°C (β-tubulin) for 15 seconds, and 72°C for 30 seconds; and then 72°C for 2 minutes. We prepared a pooled library from amplicons as previously described (M.A. Bär et al., unpub. data). We sequenced amplicons on an MiSeq instrument (Illumina) by using Illumina V3 (2 × 300 bp) sequencing chemistry for ITS2, nad-1, and nad-4, and V2 (2 × 250 bp) sequencing chemistry for ITS1 and cox-1. DNA amplicon sequencing data from this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra; BioProject no. PRJNA1131306).
We performed quality filtering of paired-end reads and primer removal by using Cutadapt version 3.2 (19) and analyzed the adaptor-trimmed paired-end reads by using DADA2 as previously described (20,21). We trimmed forward reads to 240 bp for the ITS1, ITS2, cox-1, nad-1, and nad-4 amplicons and trimmed reverse reads to 190 bp (ITS1), 220 bp (ITS2), 230 bp (cox-1), 220 bp (nad-1), and 220 bp (nad-4). We removed reads if they were <50 bp or had an expected error rate of >1 (ITS1), >3 (ITS2), >1 (cox-1), >2 (nad-1), and >2 (nad-4) nucleotides for forward reads and >2 (ITS1), >5 (ITS2), >2 (cox-1), >5 (nad-1), and >5 (nad-4) nucleotides for reverse reads. We aligned amplicon sequence variants (ASVs) to reference sequences (Appendix Table 3) by using the MAFFT tool for multiple sequence alignment (22), and we removed off-target ASVs. We performed manual correction of sequence alignments by using Geneious version 10.0.9 (Geneious, https://www.geneious.com). In the final analysis, we only included samples that had a total read depth of >1,000 mapped reads, >0.1% ASVs in the population overall, and >200 reads in an individual sample.
Phylogenetic and Population Genetic Analysis of Metabarcoding Data
We performed multiple sequence alignments for Trichuris ITS1, ITS2, cox-1, nad-1, and nad-4 reference sequences from GenBank (Appendix Table 3) and for the ASVs from amplicon sequencing by using the MAFFT tool as described. After manual correction for indels in Geneious software, we performed phylogenetic analysis by using the maximum-likelihood method. We constructed statistical parsimony haplotype networks from the corrected ASV alignments by using the pegas package (23) in R (The R Project for Statistical Computing, https://www.r-project.org). We then visualized and annotated those networks by using Cytoscape version 3.9.1 (24).
After ASV filtering and alignment, we generated haplotype distribution bar charts in R by using the ggplot2 package (25). We calculated the Shannon-Wiener index for α diversity by using the vegan package (https://github.com/vegandevs/vegan) in R and plotted each population by using ggplot2. We used pairwise t-tests to determine significant differences in the α diversity between populations. For β diversity analysis, we used a Bray-Curtis dissimilarity matrix to calculate differences in relative abundance and Jaccard distance to calculate the presence or absence of ASVs among the samples. We performed principal coordinate analysis by using the vegan package in R and generated those plots by using ggplot2. We calculated nucleotide diversity and ASV heterozygosity by using the pegas package, whereas we performed pairwise fixation index (FST) calculations and significance testing by using Arlequin version 3.5 (26) and 1,000 permutations.
Adult Worm Whole-Genome Sequencing and Analysis
We prepared adult worm sequencing libraries by using the NEBNext Ultra II FS DNA Library Prep Kit (New England Biolabs, https://www.neb.com) and measured DNA concentrations by using the Invitrogen dsDNA High Sensitivity Assay Kit and Qubit 4 Fluorometer (Thermo Fisher Scientific) and 2 μL of DNA extract as input. We checked fragment sizes by using 1% gel electrophoresis and sequence the libraries by using 2 × 150–bp paired-end chemistry on an Illumina MiSeq instrument.
After whole-genome sequencing, we used fastp (27) to filter reads below a Phred quality score of 15 and to trim adapters from paired-end reads. Next, we used GetOrganelle (28) to assemble mitochondrial genomes and extract ITS1 and ITS2 sequence data from each sample. For assembling mitochondrial genomes, we ran GetOrganelle with the flags “-R 10,” “-k 21, 45, 65, 85, 105” and a default animal mitochondrial genome database seed (-F animal_mt). We then annotated mitochondrial genomes in Geneious version 10.0.9 by using the “annotate from” function for Trichuris reference genomes from GenBank (Appendix Table 3) with a threshold of 90% similarity. For extracting ITS1 and ITS2 sequences, we used 2 T. suis sequences from GenBank (accessions nos. AM993005 and AM993010) as seeds; we used -F set to “anon” and a target size of 1,350 bp.
We downloaded additional mitochondrial genomes representing 13 described species of Trichuris, several undescribed Trichuris lineages, and Trichinella spp. outgroups from GenBank and sequences from a previously published study (Appendix Table 3) (29). We extracted protein coding sequences for the genes atp-6, cox-1, cox-2, cox-3, cytb, nad-1, nad-2, nad-3, nad-4, nad-4L, nad-5, and nad-6 from all 45 downloaded mitochondrial genomes; we aligned each sequence separately by using MAFFT. We calculated pairwise nucleotide identity by using Geneious. We used IQ-TREE 2 (30) to estimate the maximum-likelihood phylogeny from the concatenated alignment of the 12 protein-coding genes and estimated a separate substitution model per gene (using flag -m ModelFinder Plus). We assessed support for branches in the maximum-likelihood topology by using both ultrafast bootstrap (1,000 replicates; -B 1000) and a Shimodaira–Hasegawa–like approximate likelihood ratio test (–alrt 1000). We visualized the resulting maximum-likelihood phylogeny by using FigTree (https://github.com/rambaut/figtree) and rooted the tree on the branch leading to Trichinella spp.
ITS1 and ITS2 rDNA Metabarcoding of Côte d’Ivoire Samples
We amplified and sequenced a 733-bp ribosomal ITS1 region from 21 (Cȏte d’Ivoire), 34 (Laos), and 29 (Pemba Island) fecal samples and a 592-bp ITS2 region from 15 (Cȏte d’Ivoire), 26 (Laos), and 23 (Pemba Island) fecal samples. Paired-end reads were merged to produce a 461-bp sequence for ITS1 with an average mapped read depth of 17,246 (range 2,205–28,402) and a 457-bp sequence for ITS2 with an average read depth of 7,660 (range 1,172–18,027).
Figure 1
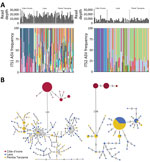
Figure 1. Frequencies and haplotype networks for ASVs of Trichuris spp. ITS1 and ITS2 in study of novel Trichuris incognitaidentified in patient fecal samples from Côte d’Ivoire and...
Many ASVs were shared between the Trichuris populations in Laos and Pemba Island, but no ASVs were shared with the Cȏte d’Ivoire population (Figure 1, panel A). Pairwise FST values were higher in Cȏte d’Ivoire when compared with either Laos and Pemba Island values than values between Laos and Pemba Island (Table). The ITS1 and ITS2 ASVs in the Cȏte d’Ivoire Trichuris population were genetically divergent from those found in the Laos and Pemba Island populations, separated by 145/461 nt difference for ITS1 and 196/457 nt differences for ITS2 (Figure 1, panel B).
Figure 2
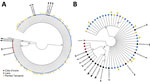
Figure 2. Phylogenetic analysis of ITS1 and ITS2 rRNA ASVs in study of novel Trichuris incognitaidentified in patient fecal samples from Côte d’Ivoire. Maximum-likelihood method was used to construct trees...
Figure 3

Figure 3. Alpha diversity of ITS1 and ITS2 rRNA amplicon sequence variants in study of novel Trichuris incognitaidentified in patient fecal samples from Côte d’Ivoire. Shannon-Wiener Index values, indicating α...
Phylogenetic analysis of Trichuris ITS1 and ITS2 ASVs from the 3 geographic regions had broadly congruent results for those 2 markers (Figure 2). All Trichuris ASVs from Cȏte d’Ivoire formed a phylogenetic clade separate from those from Laos and Pemba Island, clustering with Trichuris reference sequences from human patients in Cameroon and Uganda and from captive nonhuman primates from Italy, Czech Republic, and Uganda. This clade was more closely related to the clade containing T. suis than that containing ASVs from Laos and Pemba Island for both markers (Figure 2). Trichuris ITS2 and ITS1 ASVs from Côte d’Ivoire shared 65% (ITS1) and 80% (ITS2) identity with T. suis sequences but only 44% (ITS-1) and 56% (ITS2) identity with ASVs from Laos and Pemba Island. ASV heterozygosity and nucleotide diversity for both ITS1 and ITS2 were higher in samples from Pemba Island and Laos with much higher α diversity than in samples from Cȏte d’Ivoire (Figure 3; Appendix Table 4).
Mitochondrial Gene Metabarcoding and Population Substructuring
Species-specific primers designed by using T. trichiura reference sequences (GenBank accession nos. NC_017750, GU385218, AP017704, and KT449825) successfully generated mitochondrial cox-1, nad-1, and nad-4 amplicons for metabarcoding of most fecal samples from Laos and Pemba Island but not for any samples from Cȏte d’Ivoire, suggesting primer site sequence polymorphism of those genes in the Trichuris population of Cȏte d’Ivoire (Appendix Figure 1). Mitochondrial cox-1, nad-1, and nad-4 ASVs generated from Laos and Pemba Island fecal samples revealed high α diversity (Appendix Figures 2, 3), and all ASVs clustered phylogenetically with T. trichiura reference sequences, supporting their species identity (Appendix Figure 4). Although pairwise FST values between T. trichiura populations from Pemba Island and Laos were low (0.024 [cox-1], 0.002 [nad-1], and 0.142 [nad-4]), multidimensional metric β diversities and haplotype networks indicated some geographic substructuring between those regions, particularly for the nad-1 gene marker (Appendix Figures 5, 6).
Analysis of Complete Mitochondrial Genomes in Adult Worms from Côte d’Ivoire
Figure 4
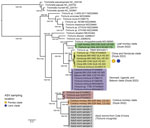
Figure 4. Phylogenetic analysis of Trichuris spp. from complete mitochondrial genome sequences in study of novel Trichuris incognitaidentified in patient fecal samples from Côte d’Ivoire. Tree was reconstructed...
The primers designed by using T. trichiura mitochondrial reference sequences did not yield amplicons from the Cȏte d’Ivoire fecal DNA samples; therefore, we extracted and assembled complete mitochondrial genomes from whole-genome sequencing data obtained from 8 adult Trichuris worms collected in a separate anthelmintic expulsion study in Cȏte d’Ivoire (M.A. Bär et al., unpub. data). Average mitochondrial genome coverage was 2,541×, and the 8 assembled mitochondrial genomes ranged from 14,253 bp to 14,663 bp (average 14,338.13 bp), slightly larger than T. trichiura reference genomes retrieved from GenBank (range 14,046–14,091; n = 6) but similar to those of T. suis (range 14,436–14,521; n = 3). Phylogenetic reconstruction of protein coding genes from Trichuris mitochondria indicated that the 8 Cȏte d’Ivoire mitochondrial genomes formed a single, well-supported clade, sister to Trichuris from Colobus monkeys (29) and more genetically distant from T. trichiura (Figure 4). Despite this phylogenetic similarity, adult worms sampled from Cȏte d’Ivoire were genetically distinct from those collected from Colobus monkeys as well as T. suis worms; pairwise nucleotide identities of DNA from Cȏte d’Ivoire were 80.2% (compared with Colobus monkeys) and 78.9% (compared with T. suis) for cox-1 and 73.1% (compared with Colobus monkey) and 71.7% (compared with T. suis) across all protein-coding genes (Appendix Table 5).
The ITS1 and ITS2 rDNA sequences recovered from the whole-genome sequences of the 8 adult worms were aligned to the ASVs generated from fecal metabarcoding. Sequence identities ranged from 93.5% to 99.8%, indicating that the same species of Trichuris was sampled in both the adult worm sequences and the fecal DNA metabarcoding (Appendix Figures 7, 8).
Co-administration of benzimidazoles with macrocyclic lactones provides much higher efficacy against T. trichiura than monotherapy with either drug class (5–10). A double-blind, randomized, controlled trial was previously conducted to compare the efficacy of an albendazole/ivermectin combination with albendazole monotherapy in Cȏte d’Ivoire, Laos, and Tanzania (12). Although the combination therapy had much higher efficacy than albendazole monotherapy in Laos and Tanzania, the efficacy in Cȏte d’Ivoire was lower and comparable to that observed for monotherapy. We used short-read metabarcoding of several taxonomic markers to compare the genetics of the Trichuris populations in fecal samples from multiple patients at each of the same 3 study sites. Analysis of the ITS1 and ITS2 ASVs revealed a genetically divergent Trichuris population in Cȏte d’Ivoire that did not share any ASVs with the samples in Laos or Pemba Island (Figure 1). Pairwise FST calculations and haplotype networks revealed substantial population differentiation in the Cȏte d’Ivoire Trichuris population.
The ASVs of both ITS1 and ITS2 rRNA DNA regions clustered into 2 distinct phylogenetic clades, clades A and B (Figure 2). All ASVs from patients in Laos and Pemba Island belonged to clade A, which corresponds to the most described reference sequences for T. trichiura in public databases (31–38). Various nomenclature designations have been used in public databases and the literature for Trichuris sequences within clade A: subclade DG (32), group 1 (33), clade DG (36), clade 2 (37), or subgroup 1 (38). In contrast, all Trichuris ASVs generated from Côte d’Ivoire patient samples fell into a second monophyletic clade B, which were phylogenetically closer to reference sequences from the porcine parasite T. suis than to the human parasite T. trichiura (Figure 2). Nevertheless, clade B is distinct from T. suis and is more closely related to a small number of reference sequences from humans and nonhuman primates previously deposited in public databases (32,33,36–38). Specifically, clade B sequences are related to 1 human patient from Cameroon, 1 human patient from Uganda (36), and several additional reference sequences from red Colobus monkeys in Uganda, captive vervet monkeys in Italy, and captive lion-tailed macaques in the Czech Republic (32,33) (Figure 2). The nomenclature previously used in the literature for this sequence group is also varied: subclade CA (32), group 3 (33), CP-GOB (36), clade 1 (37), or subgroup 5 (38).
Primers designed to amplify T. trichiura mitochondrial cox-1, nad-1 and nad-4 reference sequences consistently generated PCR amplicons from samples from Laos and Pemba Island, but not from Côte d’Ivoire. The PCR amplification failure for the Côte d’Ivoire samples is unsurprising given the genetic divergence of this Trichuris population from T. trichiura. Although some genetic substructuring in the Trichuris population was apparent in samples from Laos and Pemba Island according to mitochondrial DNA metabarcoding, all ASVs from those 2 sites clustered with human-derived T. trichiura reference sequences from public databases, including sequences from China, Japan, and Honduras, and with Trichuris sequences from captive baboons in the United States (29) (Appendix Figure 4). Together with the ITS1 and ITS2 ASV data, the ASV data indicate the albendazole/ivermectin-sensitive Trichuris populations from Laos and Pemba Islane are entirely composed of T. trichiura.
Figure 5
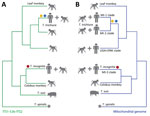
Figure 5. Schematic of phylogenetic relationships of Trichurisspp. infecting humans and nonhuman primates adapted from previously published studies. Relationships are indicated for the ribosomal ITS1-5.8S-ITS2 region (A) and the mitochondrial...
To investigate the mitochondrial phylogeny of the Trichuris sequences, we assembled the complete mitochondrial genomes from 8 adult Trichuris worms collected from an expulsion study conducted in Côte d’Ivoire (M.A. Bär et al., unpub. data). Although the mitochondrial genome sequences from Côte d’Ivoire clarify their phylogenetic relationship to other Trichuris spp. sequences, the lack of amplicon sequencing data limits our ability to assess population diversity. Phylogenetic analysis of the 8 assembled mitochondrial genomes, in conjunction with 37 previously released mitochondrial genomes (29), produced a similar outcome observed for the ITS marker analysis. Those 8 mitochondrial genomes are more closely related to Trichuris spp. from Colobus monkeys and T. suis than they are to T. trichiura (Figure 4). Despite the phylogenetic relationship of Trichuris mitochondrial genes from Côte d’Ivoire to those of Trichuris spp. from other animals, the genetic distances between lineages, shown in our phylogenetic analysis and pairwise nucleotide identity analysis (Appendix Table 5), indicate that we have sampled a new species of Trichuris. The reference genome of this Trichuris species, proposed to be T. incognita, has been characterized concurrently with this study (M.A. Bär, et al. unpub. data), and phylogenetic relationships were determined between T. trichiura, T. suis, and T. incognita (Figure 5).
The study site in Côte d’Ivoire has the longest record of consistent use of albendazole/ivermectin treatment in mass drug administration (MDA) campaigns aimed at combating lymphatic filariasis (>2 decades) (12,39). This record contrasts with that in Pemba Island, where the lymphatic filariasis MDA ended many years ago, and that in Laos, where ivermectin has not been previously used (12). It is possible that this treatment history has provided a selective advantage for T. incognita in Côte d’Ivoire because of an inherently lower sensitivity to this drug combination compared with T. trichiura.
Despite the higher (mean EPG 350 [range 110–1,151]) fecal egg counts in fecal samples from Côte d’Ivoire than those in Laos (mean EPG 147 [range 55–700]) and Pemba Island (mean EPG 202 [range 51–803]), the T. incognita population in Côte d’Ivoire had low ITS1 and ITS2 α diversity relative to the T. trichiura populations (Figure 3; Appendix Table 4). It is unlikely that a higher allelic dropout occurred for T. incognita than for T. trichiura because the primers used in this study had 100% identity with ITS1 and ITS2 from T. suis reference sequences. Low genetic diversity of the T. incognita population might indicate that this species has recently adapted to human hosts, and its presence in nonhuman primates might be consistent with this hypothesis (40,41). Alternatively, the low diversity could be from selective pressure because of the long history of albendazole/ivermectin MDA in the region.
In conclusion, we have identified T. incognita in Côte d’Ivoire, which is more closely related to T. suis than to T. trichiura and is less responsive to albendazole/ivermectin combination therapy. This work demonstrates how fecal DNA metabarcoding can be used to characterize human helminth populations at the community level. The application of fecal DNA metabarcoding by using ITS1 and ITS2, as well as other markers, will be a powerful method to investigate the geographic distribution, treatment efficacy, and control strategies for Trichuris spp. (N. Rahman et al., unpub. data, https://doi.org/10.1101/2024.07.31.605962).
Dr. Venkatesan is a research and partners manager at the Applied Genomics Centre, Kwantlen Polytechnic University in Surrey, British Colombia, Canada. Her research focuses on leveraging genomic tools to investigate infectious diseases and advance One Health initiatives.
References
- Pullan RL, Smith JL, Jasrasaria R, Brooker SJ. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit Vectors. 2014;7:37. DOIPubMedGoogle Scholar
- Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521–32. DOIPubMedGoogle Scholar
- Montresor A, Mupfasoni D, Mikhailov A, Mwinzi P, Lucianez A, Jamsheed M, et al. The global progress of soil-transmitted helminthiases control in 2020 and World Health Organization targets for 2030. PLoS Negl Trop Dis. 2020;14:
e0008505 . DOIPubMedGoogle Scholar - World Health Organization. 2030 targets for soil-transmitted helminthiases control programmes. 2020 [cited 2024 Nov 7]. https://apps.who.int/iris/handle/10665/330611
- Moser W, Schindler C, Keiser J. Efficacy of recommended drugs against soil transmitted helminths: systematic review and network meta-analysis. BMJ. 2017;358:j4307. DOIPubMedGoogle Scholar
- Belizario VY, Amarillo ME, de Leon WU, de los Reyes AE, Bugayong MG, Macatangay BJC. A comparison of the efficacy of single doses of albendazole, ivermectin, and diethylcarbamazine alone or in combinations against Ascaris and Trichuris spp. Bull World Health Organ. 2003;81:35–42.PubMedGoogle Scholar
- Ismail MM, Jayakody RL. Efficacy of albendazole and its combinations with ivermectin or diethylcarbamazine (DEC) in the treatment of Trichuris trichiura infections in Sri Lanka. Ann Trop Med Parasitol. 1999;93:501–4. DOIPubMedGoogle Scholar
- Knopp S, Mohammed KA, Speich B, Hattendorf J, Khamis IS, Khamis AN, et al. Albendazole and mebendazole administered alone or in combination with ivermectin against Trichuris trichiura: a randomized controlled trial. Clin Infect Dis. 2010;51:1420–8. DOIPubMedGoogle Scholar
- Palmeirim MS, Hürlimann E, Knopp S, Speich B, Belizario V Jr, Joseph SA, et al. Efficacy and safety of co-administered ivermectin plus albendazole for treating soil-transmitted helminths: A systematic review, meta-analysis and individual patient data analysis. PLoS Negl Trop Dis. 2018;12:
e0006458 . DOIPubMedGoogle Scholar - Matamoros G, Sánchez A, Gabrie JA, Juárez M, Ceballos L, Escalada A, et al. Efficacy and safety of albendazole and high-dose ivermectin coadministration in school-aged children infected with Trichuris trichiura in Honduras: a randomized controlled trial. Clin Infect Dis. 2021;73:1203–10. DOIPubMedGoogle Scholar
- World Health Organization. Guideline: preventive chemotherapy to control soil-transmitted helminth infections in at-risk population groups. 2017 [cited 2024 Nov 7]. https://www.who.int/publications/i/item/9789241550116
- Hürlimann E, Keller L, Patel C, Welsche S, Hattendorf J, Ali SM, et al. Efficacy and safety of co-administered ivermectin and albendazole in school-aged children and adults infected with Trichuris trichiura in Côte d’Ivoire, Laos, and Pemba Island, Tanzania: a double-blind, parallel-group, phase 3, randomised controlled trial. Lancet Infect Dis. 2022;22:123–35. DOIPubMedGoogle Scholar
- Verweij JJ, Stensvold CR. Molecular testing for clinical diagnosis and epidemiological investigations of intestinal parasitic infections. Clin Microbiol Rev. 2014;27:371–418. DOIPubMedGoogle Scholar
- Andersen LO, Röser D, Nejsum P, Nielsen HV, Stensvold CR. Is supplementary bead beating for DNA extraction from nematode eggs by use of the NucliSENS easyMag protocol necessary? J Clin Microbiol. 2013;51:1345–7. DOIPubMedGoogle Scholar
- Demeler J, Ramünke S, Wolken S, Ianiello D, Rinaldi L, Gahutu JB, et al. Discrimination of gastrointestinal nematode eggs from crude fecal egg preparations by inhibitor-resistant conventional and real-time PCR. PLoS One. 2013;8:
e61285 . DOIPubMedGoogle Scholar - Liu J, Gratz J, Amour C, Kibiki G, Becker S, Janaki L, et al. A laboratory-developed TaqMan Array Card for simultaneous detection of 19 enteropathogens. J Clin Microbiol. 2013;51:472–80. DOIPubMedGoogle Scholar
- Kaisar MMM, Brienen EAT, Djuardi Y, Sartono E, Yazdanbakhsh M, Verweij JJ, et al. Improved diagnosis of Trichuris trichiura by using a bead-beating procedure on ethanol preserved stool samples prior to DNA isolation and the performance of multiplex real-time PCR for intestinal parasites. Parasitology. 2017;144:965–74. DOIPubMedGoogle Scholar
- Avramenko RW, Redman EM, Lewis R, Yazwinski TA, Wasmuth JD, Gilleard JS. Exploring the gastrointestinal “nemabiome”: deep amplicon sequencing to quantify the species composition of parasitic nematode communities. PLoS One. 2015;10:
e0143559 . DOIPubMedGoogle Scholar - Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2. DOIGoogle Scholar
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. DOIPubMedGoogle Scholar
- Jimenez Castro PD, Venkatesan A, Redman E, Chen R, Malatesta A, Huff H, et al. Multiple drug resistance in hookworms infecting greyhound dogs in the USA. Int J Parasitol Drugs Drug Resist. 2021;17:107–17. DOIPubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Paradis E. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26:419–20. DOIPubMedGoogle Scholar
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. DOIPubMedGoogle Scholar
- Wickham H. ggplot2: elegant graphics for data analysis. Cham (Switzerland): Springer; 2016.
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7. DOIPubMedGoogle Scholar
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. DOIPubMedGoogle Scholar
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:241. DOIPubMedGoogle Scholar
- Doyle SR, Søe MJ, Nejsum P, Betson M, Cooper PJ, Peng L, et al. Population genomics of ancient and modern Trichuris trichiura. Nat Commun. 2022;13:3888. DOIPubMedGoogle Scholar
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic rra. Mol Biol Evol. 2020;37:1530–4. DOIPubMedGoogle Scholar
- Betson M, Søe MJ, Nejsum P. Human trichuriasis: whipworm genetics, phylogeny, transmission and future research directions. Curr Trop Med Rep. 2015;2:209–17. DOIGoogle Scholar
- Cavallero S, De Liberato C, Friedrich KG, Di Cave D, Masella V, D’Amelio S, et al. Genetic heterogeneity and phylogeny of Trichuris spp. from captive non-human primates based on ribosomal DNA sequence data. Infect Genet Evol. 2015;34:450–6. DOIPubMedGoogle Scholar
- Ghai RR, Simons ND, Chapman CA, Omeja PA, Davies TJ, Ting N, et al. Hidden population structure and cross-species transmission of whipworms (Trichuris sp.) in humans and non-human primates in Uganda. PLoS Negl Trop Dis. 2014;8:
e3256 . DOIPubMedGoogle Scholar - Hansen TVA, Thamsborg SM, Olsen A, Prichard RK, Nejsum P. Genetic variations in the beta-tubulin gene and the internal transcribed spacer 2 region of Trichuris species from man and baboons. Parasit Vectors. 2013;6:236. DOIPubMedGoogle Scholar
- Hawash MBF, Andersen LO, Gasser RB, Stensvold CR, Nejsum P. Mitochondrial genome analyses suggest multiple Trichuris species in humans, baboons, and pigs from different geographical regions. PLoS Negl Trop Dis. 2015;9:
e0004059 . DOIPubMedGoogle Scholar - Ravasi DF, O’Riain MJ, Davids F, Illing N. Phylogenetic evidence that two distinct Trichuris genotypes infect both humans and non-human primates. PLoS One. 2012;7:
e44187 . DOIPubMedGoogle Scholar - Rivero J, Cutillas C, Callejón R. Trichuris trichiura (Linnaeus, 1771) from human and non-human primates: morphology, biometry, host specificity, molecular characterization, and phylogeny. Front Vet Sci. 2021;7:
626120 . DOIPubMedGoogle Scholar - Xie Y, Zhao B, Hoberg EP, Li M, Zhou X, Gu X, et al. Genetic characterisation and phylogenetic status of whipworms (Trichuris spp.) from captive non-human primates in China, determined by nuclear and mitochondrial sequencing. Parasit Vectors. 2018;11:516. DOIPubMedGoogle Scholar
- Koudou BG, Kouakou MM, Ouattara AF, Yeo S, Brika P, Meite A, et al. Update on the current status of onchocerciasis in Côte d’Ivoire following 40 years of intervention: Progress and challenges. PLoS Negl Trop Dis. 2018;12:
e0006897 . DOIPubMedGoogle Scholar - Hafner MS, Nadler SA. Phylogenetic trees support the coevolution of parasites and their hosts. Nature. 1988;332:258–9. DOIPubMedGoogle Scholar
- Pérez SD, Grummer JA, Fernandes-Santos RC, José CT, Medici EP, Marcili A. Phylogenetics, patterns of genetic variation and population dynamics of Trypanosoma terrestris support both coevolution and ecological host-fitting as processes driving trypanosome evolution. Parasit Vectors. 2019;12:473. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: December 18, 2024
1Current affiliation: Kwantlen Polytechnic University, Surrey, British Columbia, Canada.
Table of Contents – Volume 31, Number 1—January 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
John Gilleard, Faculty of Veterinary Medicine, Room HSC 2557, 3330, Hospital Drive, University of Calgary, Calgary, AB T2N 4N1, Canada
Top