Volume 32, Number 6—June 2026
Dispatch
Repeated Extraneous Introductions of Cholera, Thailand, 2007–2025
Cite This Article
Citation for Media
Abstract
Genomic analyses identified 4 seventh pandemic Vibrio cholerae El Tor clades in Thailand (2007–2025). Closely related to other South Asian strains, the clades reveal that repeated cross-border introductions, rather than local persistence, drive outbreaks. Our findings highlight the importance of genomic surveillance for monitoring transmission and informing regional control strategies.
Cholera, caused by Vibrio cholerae O1, is a potentially life-threatening diarrheal disease and remains a major global public health threat (1). In recent years, large-scale outbreaks have occurred in several countries in Africa and the Middle East, including Sudan and Yemen, where conflict, population displacement, and limited access to clean water have contributed to increased transmission (2,3). South Asia, particularly the Bengal Delta, serves as a hub for genetically diverse pandemic strains and contributes to their global dissemination (4,5). Outbreak occurrence and severity are influenced by environmental and socio-economic factors, and V. cholerae can persist in aquatic environments, although long-term local persistence varies by region (6–9).
Cholera has resurged periodically in Thailand, notably from 2007, when a large-scale outbreak affected 50 provinces, peaking in northeastern regions between September and November. Analyses during 2007–2010 using pulsed field gel electrophoresis and multilocus variable-number tandem repeat analysis indicated that those outbreaks were driven by repeated introductions of V. cholerae O1, particularly in border areas (10). Independent evidence further suggested regional transmission or shared infection sources among Thailand, Laos, and Vietnam. Previous studies have revealed high clonal diversity among Thailand isolates collected during 1999–2002 (11), whereas isolates of the El Tor serotype Ogawa strain from 2007 displayed a distinct ribotype, consistent with the introduction of a new clone in northeastern Thailand. Despite those insights, understanding of the genomic relationships between historical and more recent Thailand isolates and global pandemic strains remains incomplete.
We analyzed 498 Vibrio cholerae O1 isolates from Thailand collected from 2007 through April 2025 and selected 157 representative isolates for whole-genome sequencing: 85 from 2007–2012, 52 from 2015–2016, and 20 from 2023–2025 (including 72 newly sequenced in this study). We chose our isolate pool to reflect the genetic diversity observed in the multilocus variable-number tandem-repeat analysis and to ensure variation in the collection site and isolation period (Appendix 1 Tables 1, 2). Cholera case numbers and their geographic distribution in Thailand, obtained from reports by the Division of Epidemiology (Appendix 2 Figure 1), showed a marked nationwide decline in outbreak size and frequency. From 2017 to early 2025, the annual number of cases in Thailand remained <20 (mean 7.2), and no large-scale, nationwide outbreaks occurred during that period. We included in our analyses isolates from smaller clusters near the northwestern border and sporadic cases from samples collected in late 2024 and early 2025.
We conducted a series of analyses to investigate the evolutionary dynamics of V. cholerae O1 isolates in Thailand during 2007–2025, clarifying their origins, transmission patterns, and the relationships between isolates from Thailand and those from outside the country. We sequenced genomic DNA from the isolates using an Illumina MiSeq platform (https://www.illumina.com) and mapped to the V. cholerae N16961 reference genome. For global comparison, we curated V. cholerae O1 genomes from the National Center for Biotechnology Information Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) and GenBank on the basis of published studies (Appendix 1 Table 3), ensuring balanced geographic and temporal representation. We quality-filtered data from the Sequence Read Archive using fastp software (https://github.com/opengene/fastp; -q parameter), retaining only high-quality sequences. We then identified core genome single-nucleotide polymorphisms (SNPs) using Snippy (https://github.com/tseemann/snippy), excluding regions associated with prophages (identified using PHAST [https://github.com/CshlSiepelLab/phast]), repeats (identified using NUCmer [https://github.com/nf-core/modules/tree/master/modules/nf-core/nucmer]), and recombination (identified using Gubbins [https://github.com/nickjcroucher/gubbins]). We masked these regions using VCFtools (https://github.com/vcftools/vcftools) before phylogenetic analysis. We reconstructed maximum-likelihood phylogenetic trees using RAxML software (https://cme.h-its.org/exelixis/web/software/raxml) and analyzed temporal signals using root-to-tip regression. We estimated divergence times using Bayesian molecular clock analysis. Pairwise SNP comparisons identified the 5 closest non-Thailand isolates for each outbreak cluster (Table). We used CholeraeFinder (Center for Genomic Epidemiology, https://www.genomicepidemiology.org) to detect virulence genes and mobile genetic elements. We also used Mantel tests as a complementary metric to assess broad congruence but drew the primary conclusions from the phylogenetic topologies to account for evolutionary structure.
Figure 1
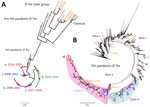
Figure 1. Maximum-likelihood phylogenetic trees of Vibrio choleraeO1 isolates reconstructed for study of repeated extraneous introductions of cholera, Thailand, 2007–2025. A) Phylogenetic relationships of 157 Thailand isolates and 11 representative...
Figure 2

Figure 2. Phylogenetic relationships, genomic features, and outbreak cluster associations of Vibrio choleraeisolates from study of repeated extraneous introductions of cholera, Thailand, 2007–2025. Black dots indicate newly sequenced Thailand isolates...
Comparative genomic analysis of 157 Thailand isolates and global genomes identified 2 lineages: the seventh pandemic El Tor (7PET; 150 isolates) and the El Tor sister group (ST75; 7 isolates) (Figure 1). The 7PET clade comprised 4 distinct groups (TH1–TH4), corresponding to 3 outbreak periods (Figure 2). Period I (2007–2012) included TH1 (clusters a and b, Ogawa, CTX-3) and TH2 (cluster c, Inaba, CTX-3), with characteristic deletions in Vibrio seventh pandemic island [VSP] II. Period II (2015–2016) included TH3 (cluster d, Ogawa), characterized by CTX-3, VPI-1ΔVC0819, ΔhlyA, and variable presence of PLE1, which varied by the geographic region of cholera cases. Period III (2024–2025) was dominated by TH4 (cluster e, Ogawa), carrying CTX-3b (ctxB7), OmpU G325D, and deletions in VSP-II. Although deletions in canonical pathogenicity islands (e.g., VSP-II, VPI) could theoretically influence fitness, we observed no obvious association with clinical severity in the study dataset.
Figure 3

Figure 3. Temporal signal and divergence dating of Thailand Vibrio choleraeO1 isolates during the seventh pandemic from study of repeated extraneous introductions of cholera, Thailand, 2007–2025. Maximum clade credibility trees...
We conducted temporal analysis (Appendix 2 Figure 2) and Bayesian molecular clock modeling (8,137 recombination-filtered SNPs) to estimate the temporal dynamics and evolutionary divergence of V. cholerae clades circulating in Thailand and worldwide (Figure 3). The clades exhibited clear evolutionary patterns over time, and the statistical analysis was well supported (effective sample size >200), indicating that the estimated divergence times were reliable. Thailand clades were closely related to other South Asia clades: TH1 corresponded to BD-2 (MAB004 and MAB006), TH2 to BD-1 (MAB001), TH3 to BD-2 (HCIS-055B, BGD133, and IDH-7956), and TH4 to BD-1.2 (DMAVC-4, -8, -17, -18, and -19). In Bangladesh, BD-1 and BD-2 have been the 2 most prominent clades during the past 2 decades, with BD-1.2 emerging more recently and responsible for a massive 2022 cholera outbreak (12).
This study confirmed that Thailand experienced multiple independent waves of cholera during 2007–2025, primarily associated with repeated introductions of V. cholerae O1 strains from South Asia. Outbreak clades in Thailand during period I (2007–2012) were most closely related to the prominent BD-1 and BD-2 clades in Bangladesh. During period II (2015–2016), 3 distinct waves emerged in both the northwestern and southern regions of Thailand, and the southern isolates exhibited multilocus variable-number tandem-repeat analysis profiles identical to strains predominant in Mandalay, Myanmar, 6 months earlier (13), suggesting direct cross-border introductions. By period III (2024–2025), most cholera cases in Thailand were imported, primarily from Myanmar, which reported >2,000 cases in 2024 (1). Of note, CTX-3b isolates in Thailand were first detected in period III and belonged to the recently emerged BD-1.2 clade, responsible for a massive 2022 cholera outbreak in Bangladesh. Although our phylogenetic data highlight repeated introductions as the primary driver of outbreaks, the potential role of long-term environmental persistence cannot be ruled out owing to a lack of systematic environmental genomic data.
Our analysis also revealed non-7PET (El Tor sister, sequence type [ST] 75) isolates: 6 of the 7 we detected lacked cholera toxin genes and 1 was the toxigenic strain MS6 previously described in a border area in 2008 (14). During 2010–2020, ST75, rather than ST69, the major seventh pandemic lineage, emerged as the dominant clonal group in China and South Africa (15).
Although the geographic spread and origins of ST75 in Thailand remain unclear, our findings demonstrate the importance of continuous genomic surveillance for elucidating cholera transmission dynamics and informing outbreak response strategies. To translate our data into public health action, implementing routine genomic sequencing at border checkpoints and establishing a real-time data sharing framework with regional partners, particularly with Myanmar, Bangladesh, and India, is essential for the early detection and mitigation of future cholera waves in the South Asia region.
Dr. Okada is an associate professor at The University of Osaka whose research interests focus on the prevention and control of diarrheal diseases caused by Vibrio cholerae and other enteropathogens of significant public health concern.
Acknowledgments
This research was supported by the Japan Initiative for Global Research Network on Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology, the Japan Agency for Medical Research and Development (grant no. 24wm0125010h0005), and the Department of Medical Sciences and Department of Disease Control, Ministry of Public Health, Thailand.
We used ChatGPT (OpenAI, https://chatgpt.com) for language refinement. All content was reviewed and verified by the authors.
References
- World Health Organization. Global situation report for cholera, 2024. Wkly Epidemiol Rec. 2025;100:347–64 https://www.who.int/publications/i/item/who-wer10036-347-364 cited 2025 Oct 1.
- Hassan IN, Ahmed S, Ahmad E, Khan A, Ashraf H. Impact of conflict on cholera epidemiology in Sudan. Lancet. 2025;406:441–2. DOIPubMedGoogle Scholar
- Weill FX, Domman D, Njamkepo E, Almesbahi AA, Naji M, Nasher SS, et al. Genomic insights into the 2016–2017 cholera epidemic in Yemen. Nature. 2019;565:230–3. DOIPubMedGoogle Scholar
- Qin C, Lypaczewski P, Sayeed MA, Cuénod A, Brinkley L, Creasy-Marrazzo A, et al. Vibrio cholerae lineage and pangenome diversity vary geographically across Bangladesh over 1 year. Microb Genom. 2025;11:
001437 . DOIPubMedGoogle Scholar - Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–5. DOIPubMedGoogle Scholar
- Shackleton D, Economou T, Memon FA, Chen A, Dutta S, Kanungo S, et al. Seasonality of cholera in Kolkata and the influence of climate. BMC Infect Dis. 2023;23:572. DOIPubMedGoogle Scholar
- Leckebusch GC, Abdussalam AF. Climate and socioeconomic influences on interannual variability of cholera in Nigeria. Health Place. 2015;34:107–17. DOIPubMedGoogle Scholar
- Colwell RR. Global climate and infectious disease: the cholera paradigm. Science. 1996;274:2025–31. DOIPubMedGoogle Scholar
- Mavian C, Paisie TK, Alam MT, Browne C, Beau De Rochars VM, Nembrini S, et al. Toxigenic Vibrio cholerae evolution and establishment of reservoirs in aquatic ecosystems. Proc Natl Acad Sci U S A. 2020;117:7897–904. DOIPubMedGoogle Scholar
- Okada K, Roobthaisong A, Nakagawa I, Hamada S, Chantaroj S. Genotypic and PFGE/MLVA analyses of Vibrio cholerae O1: geographical spread and temporal changes during the 2007–2010 cholera outbreaks in Thailand. PLoS One. 2012;7:
e30863 . DOIPubMedGoogle Scholar - Tapchaisri P, Na-Ubol M, Jaipaew J, Srimanote P, Chongsa-Nguan M, Yamasaki S, et al. Virulence genes of clinical Vibrio cholerae O1 isolates in Thailand and their ribotypes. J Infect. 2007;55:557–65. DOIPubMedGoogle Scholar
- Monir MM, Islam MT, Mazumder R, Mondal D, Nahar KS, Sultana M, et al. Genomic attributes of Vibrio cholerae O1 responsible for 2022 massive cholera outbreak in Bangladesh. Nat Commun. 2023;14:1154. DOIPubMedGoogle Scholar
- Roobthaisong A, Okada K, Htun N, Aung WW, Wongboot W, Kamjumphol W, et al. Molecular epidemiology of cholera outbreaks during the rainy season in Mandalay, Myanmar. Am J Trop Med Hyg. 2017;97:1323–8. DOIPubMedGoogle Scholar
- Okada K, Roobthaisong A, Swaddiwudhipong W, Hamada S, Chantaroj S. Vibrio cholerae O1 isolate with novel genetic background, Thailand–Myanmar. Emerg Infect Dis. 2013;19:1015–7. DOIPubMedGoogle Scholar
- Ke Z, Pang B, Yang J, Gao Y, Zhang X, Xu H, et al. Emergence and prevalence of Vibrio cholerae O1 sequence type 75 clonal complex, Fujian Province, China, 2009–2023. Emerg Infect Dis. 2025;31:1428–31. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: May 20, 2026
Table of Contents – Volume 32, Number 6—June 2026
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Kazuhisa Okada, The University of Osaka Thailand–Japan Research Collaboration Center on Emerging and Re-emerging Infections, Bldg 10, Department of Medical Sciences, Ministry of Public Health, Tiwanon Rd, Muang, Nonthaburi 11000, Thailand
Top