Volume 11, Number 8—August 2005
Research
Coxiella burnetii Genotyping
Cite This Article
Citation for Media
Abstract
Coxiella burnetii is a strict intracellular bacterium with potential as a bioterrorism agent. To characterize different isolates of C. burnetii at the molecular level, we performed multispacer sequence typing (MST). MST is based on intergenic region sequencing. These regions are potentially variable since they are subject to lower selection pressure than the adjacent genes. We screened 68 spacers in 14 isolates and selected the 10 that exhibited the most variation. These spacers were then tested in 159 additional isolates obtained from different geographic areas or different hosts or were implicated in different manifestations of human disease caused by C. burnetii. The sequence analysis yielded 30 different allelic combinations. Phylogenic analysis showed 3 major clusters. MST allows easy comparison and exchange of results obtained in different laboratories and could be a useful tool for identifying bacterial strains.
Coxiella burnetii is a strict intracellular microorganism, included in the γ subdivision of the Proteobacteria phylum (1). It is found in close association with arthropod and vertebrate hosts, and it causes Q fever in humans and animals. Cattle, goats, and sheep are the primary reservoirs of human infection. In humans, the disease may appear in 2 forms, acute and chronic (2). Acute Q fever may be asymptomatic or appear as atypical pneumonia, granulomatous hepatitis, or self-limited febrile illness. In some persons, the immune system is unable to control the infection and chronic Q fever occurs. The manifestations of chronic Q fever are endocarditis, hepatitis, osteomyelitis, or infected aortic aneurysms. C. burnetii is highly infectious by the aerosol route and can survive for long periods in the environment.
Previous studies have shown that C. burnetii isolates differed respect to their plasmid type (QpH1, QpRS, QpDG, and QpDV) (3–6), lipopolysaccharide profiles (7), and analysis of endonuclease-digested DNA separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (8) or pulsed-field gel electrophoresis (PFGE) (9–11). Differentiation was also obtained by sequence determination of the isocitrate dehydrogenase gene (12), com1 gene, and mucZ gene, which was renamed djlA when the whole genome of C. burnetii was sequenced (13,14).
Several other methods have been used to type different isolates of the same species, in particular, multilocus enzyme electrophoresis (15) and multilocus sequence typing (MLST) (16). Many bacterial species have been studied by using these approaches (17–19).
Recently, the whole genome of the C. burnetii Nine Mile strain was sequenced (14). We decided to investigate parts of the genome located between 2 open reading frames (ORFs) because they are considered potentially variable since they are subject to lower selection pressure than the adjacent genes. The 16S/23S ribosomal spacer region has been widely used to genotype bacteria (20–23). We investigated the utility of multispacer sequence typing (MST) with 173 C. burnetii isolates. After screening, we selected 10 variable spacers and showed that the combination of the different sequences allowed us to characterize 30 different genotypes. Phylogenetic analysis inferred from compiled sequences characterized 3 monophyletic groups, which could be subdivided into different clusters.
Bacterial Strains
The C. burnetii strains included in this study are listed in Table A1. All the strains were propagated on Vero cell monolayers (ATCC CRL 1587). Minimal essential medium (MEM) (Invitrogen, Cergy-Pontoise, France) supplemented with 4% fetal bovine serum (Invitrogen) and 1% L-glutamine (Invitrogen) was used for cultivation. Infected cells were maintained in a 5% CO2 atmosphere at 35°C. C. burnetii cells were harvested, pelleted, resuspended in 200 μL MEM, and mixed with 500 μL Chelex 100 20% (Bio-Rad, Ivry sur Seine, France). The preparation was boiled for 30 min, centrifuged at 10,000 × g for 30 min (24), and the supernatant containing DNA was transferred to a clean Eppendorf tube and stored at 4°C or –20°C.
Multispacer Sequence Typing
The whole genome of C. burnetii was accessible in the NCBI server (GenBank NC 002971). We kept spacers that were 300–700 bp in length. Primers were chosen in neighboring genes to allow polymerase chain reaction (PCR) amplification at 57°C and are listed in Table 1. Each PCR was carried out in a T3 Thermocycler Biometra (Biolabo, Archamps, France). Two microliters of the DNA preparation was amplified in a 50-μL reaction mixture containing 200 μmol/L of each primer, 200 μmol/L (each) dATP, dCTP, dGTP, and dTTP (Invitrogen), 1.5 U Taq DNA polymerase (Roche, Meylan, France) in 1× Taq buffer. Amplifications were carried under the following conditions: initial denaturation of 10 min at 95°C, followed by 37 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 57°C, and extension for 1 min at 72°C. PCR products were purified and sequenced as previously described (25).
PCR products were cloned in PGEM-T Easy Vector (Promega, Charbonnières, France) according to the manufacturer's instructions. Ten clones were cultivated in LB medium (USB, Cleveland, OH, USA) overnight, and PCR and sequencing were performed as described previously.
Plasmid Sequence Type, com1 Type, and djlA Type Determination
PCR for QpH1 and QpRS sequence plasmids were performed with the primers previously described QpH11/12 and QpRS01/02 (5). PCR was carried out as described for MST, except that annealing temperature was 55°C and cycle number was 35. PCR primers for QpDV and QpRS sequence plasmid amplification were chosen after comparison of the entire sequence of the 2 plasmids. The primers were QpDV1f and QpDV1r. PCR amplification was carried out at 63°C for 30 cycles. PCR was performed as previously described for com1 and djlA (13) (Table A2).
Statistical analyses were performed by using the chi-square test in the program EpiInfo 6 (26). The spacer sequences were compiled and aligned by using the multisequence alignment program ClustalX (1.8). The phylogenetic relationships between the C. burnetii isolates were determined by using Mega version 2.0 (27). A matrix of pairwise differences in allele profiles was constructed, and the similarities between the allelic profiles of the isolates were assessed by cluster analysis using the unweighted pair-group method with arithmetic mean (UPGMA). Another analysis of the results was performed by using the BURST algorithm (http://www.mlst.net), which defines clonal complexes in which every isolate shares at least 5 identical alleles with at least 1 other isolate (Cox2, Cox5, Cox18, Cox20, Cox37, Cox56, and Cox57 were kept for the analysis) and characterizes ancestral genotypes. C. burnetii MST database was entered at the following website: http://ifr48.timone.univ-mrs.fr, and ST determination by sequence comparison is possible at this site.
Choice of Spacers for Typing and Analysis by MST
Initially 14 isolates were chosen to test the genetic diversity of the spacers: Nine Mile, Priscilla, Q212, Heizberg, Brasov, Dog ut Ad, CB15, CB20, CB26, CB28, CB33, CB35, CB114, and CB115. We chose 68 spacers, but we retained only 51 spacers for which PCR amplification was obtained for all the isolates. We kept 10 spacers (Cox2, Cox5, Cox18, Cox20, Cox22, Cox37, Cox51, Cox56, Cox57, and Cox61) (Table 1) because they were representative of the results found when we analyzed the entire test set of 51 spacers. For each spacer, the number of variable sites in the sequences was determined, and the percentage of variability was calculated. They were, respectively, 1.1, 1.4, 1.9, 0.7, 2.3, 1.2, 1.4, 2.5, 1.7, and 2.1. We kept Cox18, Cox22, Cox51, Cox56, Cox57, and Cox61 because the percentage of variability in these spacers was high compared with the other spacers. We kept Cox2, Cox5, Cox20, and Cox37 because they allowed the characterization of CB35, CB15, CB26 and CB28, and Nine Mile respectively. To test the reliability of the spacers we kept, chi-square value was determined by using the value of 1% as the threshold value. The Fisher value was found to be statistically significant (9 × 10–4). We then added 159 other isolates. Sequences were obtained for all the isolates with spacers Cox2, Cox18, Cox20, Cox22, Cox37, Cox51, and Cox57. Mixed sequences were obtained with the isolate Poker Cat with spacers Cox5, Cox56, and Cox61. We cloned the PCR products and showed that several sequences were present after PCR amplification, including insertions or deletions. Allele distribution of the different gene spacers are described in Table 2. Each of the different sequences in a locus defined a distinct genotype, even if it differed from the others by only a single nucleotide. Thirty different sequence types (STs) were identified by using MST.
The nucleotide sequence accession numbers are noted in Table A3. Accession numbers for Poker Cat isolate clones are, respectively, AY619726, AY619728, and AY619729, and AY619721 for Cox5, Cox56, and Cox61.
Computer Analysis of MST Data
Figure
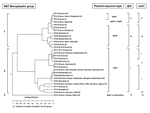
Figure. . Dendrogram of the genetic relatedness among the 30 different sequence types defined by multispacer sequence type (MST) analysis. The dendrogram was constructed by unweighted pair-group method with arithmetic mean. Plasmid...
The dendrogram in the Figure was constructed from a matrix of pairwise allelic differences between the compiled sequences of the 30 STs. We identified 3 monophyletic groups within the tree. The first group, representing 13 different STs, included isolates from France, Spain, Russia, Kyrgyzstan, Namibia, Kazakhstan, Ukraine, Uzbekistan, and the United States. It was divided in 2 subgroups. The first one included 36 isolates representing 8 different STs (ST1 to ST7 and ST30). Nineteen were represented by ST1. The second subgroup included 39 isolates which represented 5 different STs (ST8, ST9, ST10, ST26, and ST28). Twenty-eight were represented by ST8.
The second group included isolates from Europe (France, Germany, Switzerland, Romania, Italy, Greece, Austria, Slovakia), the United States, Russia, Africa (Central Africa and Senegal), and Asia (Kazakhstan, Uzbekistan, Mongolia, and Japan). It was divided into 4 subgroups. The first one included 26 isolates, which represented 7 different STs (ST11, ST12, ST13, ST14, ST15, ST24, and ST27). The second subgroup included 34 isolates that were included in ST18, ST22, ST23, ST25, and ST29 groups. The third subgroup included 18 isolates (ST16 and ST17), and the fourth subgroup included 10 isolates (ST19 and ST20).
The third group consisted of only 1 ST, ST21, and included the 7 Canadian isolates, 2 isolates from France (CB4 and CB7), and 1 isolate from the United States (Scurry). The clusters determined by the BURST algorithm were consistent with those determined by the phylogenetic analysis. Five groups were defined. The first one included ST1 to ST7; the putative ancestral genotype in this group was ST1. ST8 (putative ancestral genotype), ST9, ST10, ST26, and ST28 were included in the second group; ST11, ST12 (putative ancestral genotype), ST13, ST14, ST15, and ST24 in the third group, ST16 and ST17 in the fourth group; and ST18 (putative ancestral genotype), ST22, ST23, ST25, and ST29 in the fifth group. ST19, ST20, ST21, and ST30 were considered as singletons.
Sequence Type Determination and Correlation with Pathology
In the monophyletic group 1, the sequence of plasmid QpRS was found for isolates included in ST4, ST5, ST6, ST7, ST8, ST9, ST10, ST26, ST28, and ST30. The QpDV plasmid sequence was amplified for isolates included in ST1, ST2, ST3, and ST4. In the monophyletic group 2, the QpH1 plasmid sequence was found in all the isolates. In the monophyletic group 3, the QpH1 plasmid sequence or none of the searched plasmid sequences was detected. Sequence comparison of djlA generated 4 different groups. Group I included all STs included in the monophyletic group 2 defined by MST analysis. Group II included ST1, ST2, ST3, and ST4. Group III included ST5, ST6, ST7, ST30, ST26, ST28, ST8, ST9, and ST10. Group IV corresponded to ST21. Com1 sequence comparison generated 6 different groups. Group I included all the STs included in the monophyletic group 2 defined by MST analysis except ST14 (group V) and ST20 (groupVI). Group II included ST1, ST2, ST3, and ST4. Group III included ST5, ST6, ST7, ST30, ST26, ST28, ST8, ST9, and ST10. Group IV corresponded to ST21. When com1 typing was used, only 1 strain was not in accordance with MST typing results. This strain, CB95, was included in ST8 but exhibited a group II com1 sequence.
QpDV plasmid presence in human isolates was correlated with the acute form of the disease (p = 2 × 10–7), and QpRS plasmid presence was correlated with the chronic form of the disease (p = 2 × 10–4). The acute form of the disease was correlated with ST1 (p = 10–3), ST4 (p = 7 × 10–4) ST16 (p = 3 × 10–3), ST18 (p = 10–2), and the chronic form of the disease was correlated with ST8 (p = 2 × 10–3).
Modifications in ORFs Surrounding Studied Spacers
As primers were chosen in ORFs surrounding the studied spacers, mutations, deletions, or insertions were noted in the protein sequences. Mutations were noted in the hypothetical protein (gi29653385) for ST11; in the hypothetical protein (gi29653385) for ST9 and ST26; in entericin (gi29653446) for ST20, in ribonuclease H (gi29653667) in ST1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 21, 26, 28, and 30; in amino acid permease family protein (gi29653908) in ST28; in hypothetical protein (gi29654047) in ST1, 2, 4, 5, 6, 7, 8, 9, 10, 26, 28, and 30. In CB118 (ST3), a stop codon appeared which shortened the length of the ORF. Mutations were noted in uridine kinase (gi29654198) in ST18, ST22, ST23, ST25, and ST29; in ompA-like transmembrane domain protein (gi29654257), in ST20; in rhodanese-like domain protein (gi29654263) in ST20 (the protein was longer by 2 amino acids); in dioxygenase (gi29654325) in ST21 and ST22; in hypothetical protein (gi29732244), in ST17.
Insertions or deletions were noted in hypothetical protein (gi29653386) in ST5, 6, and 7; in hypothetical protein (gi29653755) in ST1 and ST3 (insertion of a base G in the DNA sequence made the protein sequence longer of 22 amino acids); in the amino acid permease family protein (gi29653772) in ST8, 9, and 10 (deletion of a base A in the DNA sequence made the protein sequence longer of 24 amino acids); in ompA-like transmembrane domain protein (gi29654257) in ST11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 22, 23, 24, 25, 27, and 29.
Q fever in humans and animals, caused by C. burnetii, is found worldwide. In humans, it causes a variety of diseases such as acute flulike illness, pneumonia, hepatitis, and chronic endocarditis. In animals, C. burnetii is found in the reproductive system, both uterus and mammary glands and may cause abortion or infertility.
Molecular methods are now almost universally used to characterize strains and to determine the relatedness between isolates causing diseases in different contexts. The most discriminative approach used for C. burnetii isolates until this study was PFGE. Twenty different restriction patterns were distinguished after NotI restriction of total C. burnetii DNA and PFGE (11). Comparison of PFGE profiles is sometimes difficult because good separation of the different fragments is required. For example, the isolate Heizberg was classified in group 1 by Thiele et al. (10) and in group 2 by Jäger et al. (11). This fact highlights the difficulty of comparing results obtained by this technique. Moreover, in some species, rapid genomic rearrangements occur because of repeats or insertion sequences, so even if isolates descended from a common ancestor that arose several decades ago, they may not readily be seen to be minor variants of the same clone. In these cases, PFGE does not contribute to tracing of isolates. The great advantage of MST over PFGE as a typing method is the lack of ambiguity and the portability of sequence data, which allow results from different laboratories to be compared without exchanging strains. This work is the first to include so many isolates in a rigorous examination of molecular epidemiology. The study of this bank of sequences will contribute to understanding the propagation mode of the bacteria as variations accumulate relatively slowly, thus making it an ideal tool for global epidemiology. For example, in ST16 we characterized isolates that were obtained from 1935 (Nine Mile) to 1991 (CB25).
Most of the French isolates were included in monophyletic group 1. Nineteen were included in ST1, and 24 were included in ST8. Thus, an isolate has a geographic distribution even if genetic modifications appear (insertions, deletions or mutations) over time, giving rise to a new ST that is related to the ancestor isolate. This fact was highlighted when the analysis of the STs was performed by using the BURST algorithm. ST1 and ST8 were described as the ancestral genotypes and for example, ST9 and ST10 corresponded to SLVs of ST8 (isolates that differ at only 1 of the 7 loci) and ST26 and ST28 corresponded to DLVs of ST8 (double locus variants). But some types were not delineated on the basis of geographic origin because they were isolated from different parts of the world. This distribution in distant countries is likely related to movements of infected patients, animals, or ticks. This is particularly true for ST16 isolates that were encountered on 4 different continents, America, Europe, Asia, and Africa. The homology of the Canadian isolates from Nova Scotia should be noted. Q fever is just as endemic in Nova Scotia as in France. This may indicate rapid and recent spreading of a single strain. The association between ST21 and Canada is significant as tested with the chi-square test with a Fisher value <10–8. Notably, patient CB115, who had Q fever endocarditis, was living in Edmonton, Alberta (≈3,000 miles from Nova Scotia) when this illness was diagnosed. He grew up in Nova Scotia, and the molecular epidemiologic findings show that he acquired his disease there. Q fever is uncommon in Alberta. Most of the STs are found in Europe. A sample bias could exist as most of the isolates tested were from this continent, but the results obtained may also indicate that C. burnetii originated from the Old World and spread later in the New World, excluding New Zealand.
Concordant results were found when MST was compared with com1 and djlA sequences comparison (Figure) However MST was more discriminant. Plasmid profile investigation of C. burnetii detected 4 different plasmids QpH1, QpRS, QpDV, and QpDG and 1 group of plasmidless isolates. QpH1 was first found in the Nine Mile tick isolate (28). QpRS was first found in the goat isolate Priscilla (29). QpDG was described from isolates obtained from feral rodents near Dugway, Utah (8). QpDV was found in French and Russian isolates (5,6). Another not-well-characterized plasmid type was described in China (30). The existence of a plasmidless C. burnetii isolate, Scurry Q217 was described (31), but a chromosomally integrated plasmid-homologous DNA fragment was found in this isolate by hybridization (32,33). Plasmid type sequence detection was also correlated with MST. Group 2 included isolates that PCR amplification found to be positive with primers specific for QpH1. Group 3 included 3 isolates, 2 from France (CB4 and CB7) and 1 from Nova Scotia (Poker Cat), in which plasmid sequence type of QpH1 was detected. No such sequence was detected in the other isolates of Nova Scotia origin included in group 3. Group 1 included isolates that were positive by PCR amplification with primers specific for QpRS (47/77). QpDV plasmid was described in isolates from France, Spain, Ukraine, and Kyrgyzstan. In fact, regions shared by QpH1, QpRS, and QpDV were termed "core plasmid sequences" and encompassed 25 kb. QpH1, QpRS, and QpDV are, respectively, 37 kb, 39 kb, and 33 kb in size. Integrated sequences in American isolate represent 18 kb. Differences in plasmid size and sequence can be explained by notable sequence rearrangements, such as deletions, insertions, or duplications, because several repeat sequences have been identified through which such rearrangements might have occurred. For CB13, we were able to characterize sequences for plasmids QpH1 and QpDV, which can be caused by several situations: this isolate may have 1) 2 different plasmids, 2) a QpH1 plasmid and sequences of QpDV integrated in the chromosome, or 3) a new plasmid that arose from combination of QpH1 and QpDV. All these hypotheses are in agreement with the presence of QpH1 plasmid in the ancestor of C. burnetii isolates. This plasmid was lost by some of them (monophyletic group 3) but genetic information of crucial importance for the organism was integrated in the chromosome. For other isolates, QpH1 plasmid evolved to QpRS plasmid, in some isolates QpRS plasmid evolved to QpDV plasmid.
This study showed a correlation between QpDV and acute infections, between QpRS and chronic infections, and an association between some genotypes and disease type. A bias in sampling exists since acute disease is 20 times more frequent than chronic disease, but in this study, most of the human isolates were from chronic disease patients, and the isolates from acute infections were mainly obtained from France. These facts reflect the difficulty in isolating the bacteria. A genomic typing method such as MST could be applied directly to samples to obtain a more precise idea of how C. burnetii is spreading in the environment and the pathogenetic implications in acute and chronic forms of Q fever.
Comparison of DNA sequences is the best approach to investigate bacterial evolution. MLST in association with BURST analysis has been used to type isolates of many species. But this method is useful only if housekeeping gene diversity exists in the studied species. For example, in the species Yersinia pestis no diversity was found in the housekeeping genes studied (34). With the MST approach, differentiation of the 3 biovars Antiqua, Medievalis, and Orientalis was possible (25), which shows that the discriminatory power of MST is higher than that of MLST and is comparable to that of tandem repeats analysis (35). Low variability was found in C. burnetii housekeeping genes such as 16S rRNA (36) and rpoB (37). MST is the first method that allows a rapid and reliable typing of C. burnetii isolates during investigations of outbreaks by sequencing the PCR product obtained from the 10 spacers described. We did not test isolates from Australia and only 8 from the United States. Two isolates from Africa (Namibia and CB119) were considered as singletons in the BURST analysis denoting lack of closely related isolates. In the future, isolates that were not available in our laboratory during this study must be tested so the missing links in our phylogenetic analysis can be determined. The constitution of a database in a website will allow isolates from all the countries in the world to be compared and increase understanding of the propagation of the isolates of C. burnetii.
Dr. Glazunova obtained her doctorate in life science at the Russian Medical State University (Moscow) and Irkutsk State University. She is currently a postdoctoral researcher at the Medical Faculty of Marseille. Her specialty is the molecular identification of bacteria.
Acknowledgments
We are grateful to Marie-Laure Birg and Jean-Yves Patrice for their technical assistance in culture of C. burnetii isolates.
This work was supported by a grant from the French Ministry of Research (ACI Microbiologie 2003) and a grant of the Pasteur Institute (2003-11).
References
- Weisburg WG, Dobson ME, Samuel JE, Dasch GA, Mallavia LP, Baca O, Phylogenetic diversity of the Rickettsiae. J Bacteriol. 1989;171:4202–6.PubMedGoogle Scholar
- Marrie TJ. Principles and practice of infectious diseases, 3rd edition. In: Mandel GL, Douglas RGJ, Bennett JE, editors. Coxiella burnetii (Q fever). New York: Churchill Livingstone; 1990. p. 1472–6.
- Stein A, Raoult D. Lack of pathotype specific gene in human Coxiella burnetii isolates. Microb Pathog. 1993;15:177–85. DOIPubMedGoogle Scholar
- Willems H, Thiele D, Krauss H. Plasmid differentiation and detection of Coxiella burnetii in clinical samples. Eur J Epidemiol. 1993;9:411–8. DOIPubMedGoogle Scholar
- Thiele D, Willems H. Is plasmid based differentiation of Coxiella burnetii in 'acute' and 'chronic' isolates still valid? Eur J Epidemiol. 1994;10:427–34. DOIPubMedGoogle Scholar
- Valkova D, Kazar J. A new plasmid (QpDV) common to Coxiella burnetii isolates associated with acute and chronic Q fever. FEMS Microbiol Lett. 1995;125:275–80. DOIPubMedGoogle Scholar
- Hackstadt T. Antigenic variation in the phase I lipopolysaccharide of Coxiella burnetii isolates. Infect Immun. 1986;52:337–40.PubMedGoogle Scholar
- Hendrix LR, Samuel JE, Mallavia LP. Differentiation of Coxiella burnetii isolates by analysis of restriction-endonuclease-digested DNA separated by SDS-PAGE. J Gen Microbiol. 1991;137:2697–6.PubMedGoogle Scholar
- Heinzen R, Stiegler GL, Whiting LL, Schmitt SA, Mallavia LP, Frazier ME. Use of pulsed field gel electrophoresis to differentiate Coxiella burnetii strains. In: Hechemy KE, Paretsky D, Walker DH, Mallavia, LP, editors. Rickettsiology: current issues and perspectives. Ann N Y Acad Sci. 1990;590:504–13. DOIPubMedGoogle Scholar
- Thiele D, Willems H, Köpf G, Krauss H. Polymorphism in DNA restriction patterns of Coxiella burnetii isolates investigated by pulsed field gel electrophoresis and image analysis. Eur J Epidemiol. 1993;9:419–25. DOIPubMedGoogle Scholar
- Jäger C, Willems H, Thiele D, Baljer G. Molecular characterization of Coxiella burnetii isolates. Epidemiol Infect. 1998;120:157–64. DOIPubMedGoogle Scholar
- Nguyen SV, Hirai K. Differentiation of Coxiella burnetii isolates by sequence determination and PCR-restriction fragment length polymorphism analysis of isocitrate dehydrogenase gene. FEMS Microbiol Lett. 1999;180:249–54. DOIPubMedGoogle Scholar
- Sekeyova Z, Roux V, Raoult D. Intraspecies diversity of Coxiella burnetii as revealed by com1 and mucZ sequence comparison. FEMS Microbiol Lett. 1999;180:61–7. DOIPubMedGoogle Scholar
- Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci U S A. 2003;100:5455–60. DOIPubMedGoogle Scholar
- Selander RK, Caugant DA, Ochman H, Musser JM, Gilmour MN, Whittam TS. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl Environ Microbiol. 1986;51:873–84.PubMedGoogle Scholar
- Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. DOIPubMedGoogle Scholar
- Enright MC, Spratt BG. Multilocus sequence typing. Trends Microbiol. 1999;7:482–8. DOIPubMedGoogle Scholar
- Schloter M, Lebuhn M, Heulin T, Hartmann A. Ecology and evolution of bacterial microdiversity. FEMS Microbiol Rev. 2000;24:647–60. DOIPubMedGoogle Scholar
- Urwin R, Maiden CJ. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;11:479–87. DOIPubMedGoogle Scholar
- Bes M, Slim LS, Becharnia F, Meugnier H, Vandenesch F, Etienne J, Population diversity of Staphylococcus intermedius isolates from various host species: Typing by 16S-23S intergenic ribosomal DNA spacer polymorphism analysis. J Clin Microbiol. 2002;40:2275–7. DOIPubMedGoogle Scholar
- Daffonchio D, Cherif A, Brusetti L, Rizzi A, Mora D, Boudabous A, Nature of polymorphisms in 16S-23S rRNA gene intergenic transcribed spacer fingerprinting of Bacillus and related genera. Appl Environ Microbiol. 2003;69:5128–37. DOIPubMedGoogle Scholar
- Hassan AA, Khan IU, Abdulmawjood A, Lammler C. Inter- and intraspecies variations of the 16S-23S rDNA intergenic spacer region of various streptococcal species. Syst Appl Microbiol. 2003;26:97–103. DOIPubMedGoogle Scholar
- Roux V, Raoult D. Inter- and intraspecies identification of Bartonella (Rochalimaea) species. J Clin Microbiol. 1995;33:1573–9.PubMedGoogle Scholar
- Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10:506–13.PubMedGoogle Scholar
- Drancourt M, Roux V, Dang LV, Tran-Hung L, Castex D, Chenal-Francisque V, Genotyping, Orientalis-like Yersinia pestis, and plague pandemics. Emerg Infect Dis. 2004;10:1585–92.PubMedGoogle Scholar
- Dean AG, Dean JA, Coulombier D, Brendel KA, Smith DC, Burton AH, Epi Info Version 6: a word processing, database, and statistics program for epidemiology on microcomputers. Atlanta: Centers for Disease Control and Prevention; 1994.
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;17:1244–5. DOIPubMedGoogle Scholar
- Samuel JE, Frazier ME, Kahn ML, Thomashow LS, Mallavia LP. Isolation and characterization of a plasmid from phase I Coxiella burnetii. Infect Immun. 1983;41:488–93.PubMedGoogle Scholar
- Samuel JE, Frazier ME, Mallavia LP. Correlation of plasmid type and disease caused by Coxiella burnetii. Infect Immun. 1985;49:775–9.PubMedGoogle Scholar
- Ning Z, Shu-Rong Y, Guo Quan Y, Xue Z. Molecular characterization of cloned variants of Coxiella burnetii isolated in China. Acta Virol. 1992;36:173–83.PubMedGoogle Scholar
- Savinelli EA, Mallavia LP. Comparison of Coxiella burnetii plasmids to homologouschromosomal sequences present in all plasmidless endocarditis-causing isolates. Ann N Y Acad Sci. 1990;590:523–33. DOIPubMedGoogle Scholar
- Willems H, Ritter M, Jäger C, Thiele D. Plasmid-homologous sequences in the chromosome of plasmidless Coxiella burnetii Scurry Q217. J Bacteriol. 1992;36:3293–7.PubMedGoogle Scholar
- Lautenschläger S, Willems H, Jäger C, Baljer G. Sequencing of the cryptic plasmid QpRS from Coxiella burnetii. Plasmid. 2000;44:85–8. DOIPubMedGoogle Scholar
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A. 1999;96:14043–8. DOIPubMedGoogle Scholar
- Pourcel C, André-Mazeaud F, Neubauer H, Ramisse F, Vergnaud G. Tandem repeats analysis for the high resolution phylogenetic analysis of Yersinia pestis. BMC Microbiol. 2004;4:22. DOIPubMedGoogle Scholar
- Stein A, Saunders NA, Taylor AG, Raoult D. Phylogenic homogeneity of Coxiella burnetii strains as determinated by 16S ribosomal RNA sequencing. FEMS Microbiol Lett. 1993;113:339–44. DOIPubMedGoogle Scholar
- Mollet C, Drancourt M, Raoult D. Determination of Coxiella burnetii rpoB sequence and its use for phylogenetic analysis. Gene. 1998;207:97–103. DOIPubMedGoogle Scholar
Figure
Tables
Cite This Article1Dr. Glazunova and Dr. Roux contributed equally to this work.
Table of Contents – Volume 11, Number 8—August 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Didier Raoult, Unité des Rickettsies, Faculté de Médecine, CNRS UMR 6020, IFR48, 27 Boulevard Jean Moulin, 13385 Marseille, Cedex 05, France; fax: 33-491-32-03 90.
Top