Volume 18, Number 1—January 2012
Research
Assessing Prion Infectivity of Human Urine in Sporadic Creutzfeldt-Jakob Disease
Cite This Article
Citation for Media
Abstract
Prion diseases are neurodegenerative conditions associated with a misfolded and infectious protein, scrapie prion protein (PrPSc). PrPSc propagate prion diseases within and between species and thus pose risks to public health. Prion infectivity or PrPSc presence has been demonstrated in urine of experimentally infected animals, but there are no recent studies of urine from patients with Creutzfeldt-Jakob disease (CJD). We performed bioassays in transgenic mice expressing human PrP to assess prion infectivity in urine from patients affected by a common subtype of sporadic CJD, sCJDMM1. We tested raw urine and 100-fold concentrated and dialyzed urine and assessed the sensitivity of the bioassay along with the effect of concentration and dialysis on prion infectivity. Intracerebral inoculation of transgenic mice with urine from 3 sCJDMM1 patients failed to demonstrate prion disease transmission, indicating that prion infectivity in urine from sCJDMM1 patients is either not present or is <0.38 infectious units/mL.
Prion diseases, a group of neurodegenerative disorders affecting humans and animals, have received considerable attention largely because of their intriguing pathogenetic mechanism and the threat they pose to public health because of their insidious infectivity. Despite their heterogeneity, all classic prion diseases are characterized by the presence of an abnormal isoform of the normal cellular prion protein (PrPC), which predominantly accumulates in the central nervous system (1). The abnormal isoform, identified as scrapie PrP or PrPSc, is thought to form from a posttranslational change in conformation of PrPC. Prion diseases can also be transmitted by an infectious mechanism because exogenous PrPSc can impose its conformation on to the host’s PrPC through a PrPSc-templated conversion process (1).
The most common form of human prion disease, Creutzfeldt-Jakob disease (CJD), can be sporadic, inherited, or acquired by infection; the sporadic form alone accounts for the great majority of all the cases of CJD (2). Following protease digestion, the unglycosylated form of PrPSc exhibits the electrophoretic mobilities of either 21 kDa or 19 kDa (3). These two PrPSc isoforms, named PrPSc types 1 and 2, respectively, along with the methionine/valine polymorphism at codon 129 of the PrP gene, led to the current classification of sporadic CJD (sCJD) in 5 phenotypically distinct subtypes (2,3). The sCJDMM1 subtype (affecting persons homozygous for methionine at codon 129 and carrying the PrPSc type 1) accounts for ≈70% of all cases of sCJD and unquestionably is the most prevalent type of human prion disease (2,3).
PrPSc is generally considered the major, if not the sole, component of the infectious agent in prion diseases (1,4,5). The presence of PrPSc, prion infectivity, or both has been found in several tissues and organs outside the central nervous system in prion-affected humans and animals (6–10). These findings have caused mounting concerns regarding the risk of human transmission of the disease from a variety of sources, including the consumption of prion contaminated meat and other animal products, the use of contaminated surgical instruments and medicinal products, and the exposure to waste from infected humans and animals. In this context, body fluids such as saliva, milk, and urine have received particular attention as they may support horizontal transmission and environmental contamination, which in turn may contribute to the propagation of ovine scrapie and chronic wasting disease (CWD) of cervids. Indeed, saliva has been reported to be a source of prion in scrapie-infected sheep and CWD-infected deer (11–14), whereas milk has been found to contain prions in scrapie infected sheep (15,16).
The detection of PrPSc by immunoblotting has been previously reported in urine of prion-affected hamsters and humans (17). However, this observation has not been confirmed in 3 subsequent studies, which instead have suggested that the original immunoblot finding resulted from nonspecific cross-reaction either with contaminating bacterial proteins (18) or urinary IgG fragments (19,20). Recently, prion infectivity has been detected in urine from experimentally prion-infected animals, including hamsters (21,22), deer (13), and mice in association with lymphocytic nephritis (23). However, infectivity in urine from naturally prion-affected animals has never been reported.
It is difficult to extrapolate the animal data on urine infectivity to human urine, especially for sCJD, because this form of prion disease is believed to start spontaneously in the brain rather than being caused by exogenous infection. Nevertheless, the possibility that PrPSc is indeed present in urine of sCJD patients exists because small quantities of PrPSc have been identified in peripheral organs of sCJD patients (6,7,10). Furthermore, we have recently shown that normal urine contains discrete amounts of a C-terminal fragment of PrP, matching the so-called C1 fragment but not full-length PrP (24). Although C1 may not be a good substrate for PrPSc replication, and its conversion to PrPSc has never been reported, the possibility of conversion may not be ruled out (25,26).
The presence of prions in urine of CJD patients would obviously pose serious risks relating to the medicinal use of urine-extracted proteins, hormones, and urokinase as well as collection and disposal of patient urine. Indeed, it has recently been reported that fragments of PrP, consistent with the urine PrP present in normal urine described above, co-purify with urine-derived gonadotropins (24,27). This finding has prompted the claim that PrPSc may also co-purify with urinary gonadotropins (27).
With the exception of early failed attempts to transmit prion disease to rodents and nonhuman primates with urine from CJD patients (28,29), no investigation on prion infectivity of human urine has been reported. We searched for prion infectivity in urine obtained from patients with sCJDMM1, the most common form of sCJD, by bioassay that used transgenic (Tg) mice expressing human PrP (30).
Patients
Samples from 4 patients (patients 1–4) affected by typical sCJDMM1 that was histologically and immunochemically confirmed (with no clinical signs of inflammatory kidney disease) were provided by the National Prion Disease Pathology Surveillance Center. The patients were 54, 68, 60, and 69 years old with disease durations of ≈2, 2, 3, and 2 months, respectively (2). Approvals from the Institutional Review Board and the Institutional Animal Care and Use Committee were obtained.
Urine
Urine samples from sCJDMM1 patients 1–3 collected 2 weeks, 1 month, and 1 week before death, and from 3 healthy controls, were concentrated 100-fold by ultrafiltration with Millipore Centricon Plus 70 (Millipore, Billerica, MA, USA) (10-kDa cutoff) by using a Beckman (Miami, FL, USA) centrifuge at 3,000 × g. Concentrated urine was dialyzed against phosphate-buffered saline (PBS) (4 L each time) at 4°C by using the Pierce Slide-A-Lyzer cassette (Thermo Fisher Scientific, Inc., Rockford, IL, USA) (10-kDa cutoff) with 2 additional changes over 2 days.
Brain Homogenates and Microsomal Fractions
Brain homogenates (BH) from inoculated mice (10%, wt/vol) were prepared at 4°C in PBS cleared by centrifugation at 1, 000 × g for 5 min. Microsomal fractions (MF) were prepared from the frontal cortex of the sCJDMM1 patients according to Reichl et al. (31). BH (10%, wt/vol) in PBS were centrifuged at 700 × g for 10 min at 4°C. The supernatant was collected and the pellet, resuspended in PBS at 30% (wt/vol), was centrifuged as above. This supernatant, pooled with the previous one, was recentrifuged at 10,000 × g for 7 min at 4°C. The pellet was discarded and the supernatant was centrifuged at 100,000 × g for 1 h at 4°C. The final pellet, representing the MF, was resuspended in PBS at 10% (wt/vol). Transgenic mice expressed full-length human PrP-129M at wild type level in mouse PrP null background, Tg(HuPrP-129M)Prnp0/0 (Tg40) (30).
Effect of Concentrated and Dialyzed Urine on Prion Infectivity
MF of sCJDMM1 patient 4 was spiked in 100× concentrated and dialyzed normal urine. The sample was 10-fold serially diluted and intracerebrally inoculated into Tg40 mice.
Determination of Infectivity Loss during Concentration and Dialysis
MF (100 µL) of sCJDMM1 patient 3 was spiked into normal urine (100 mL) either before or after 100× concentration and dialysis. Subsequently, 30 µL of the differently processed urine samples, both containing an equivalent amount of 3 µL of MF, were intracerebrally injected into Tg40 mice. The infectivity titers of the 2 differently processed MF spiked urines were compared on the basis of their mean incubation times in Tg40 mice.
Inoculation
Thirty microliters of MF from sCJDMM1 patients 1–4 suspended in PBS or in 100× concentrated and dialyzed normal urine were intracerebrally injected into Tg40 mice (Table 1, Table 2, Table 3, Table 4). Infectivity of urine from 3 sCJDMM1 patients and 3 healthy subjects was assayed as above by inoculation of 30 µL of 100× concentrated and dialyzed urine; raw urine from sCJDMM1 patient 1 was also similarly bioassayed in 33 Tg40 mice. Mice were euthanized within 3 days of becoming symptomatic.
Infectivity Titration
Figure 1
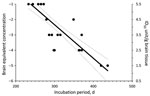
Figure 1. Dose-incubation period curve of brain microsomal fraction from sporadic Creutzfeldt-Jakob disease MM1 (patient 1) intracerebrally injected into Tg40 mice. Each solid circle represents the incubation time for single animal (x-axis) at...
Infectivity in MF from sCJDMM1 patients 1 and 4, the latter spiked and diluted in 100× concentrated and dialyzed normal urine, was estimated by endpoint dilution by using probit regression analysis (STATA version 8.2 software; StataCorp LP, College Station, TX, USA). Incubation periods (Figure 1, x-axis) of each mouse injected with known infectivity titers (Figure 1, y-axis) of sCJDMM1 patient 1, were plotted and the experimental points fitted either by linear or segmental linear regression curves (GraphPad Prism version 5.0b for Macintosh; GraphPad Software, San Diego, CA, USA), and the 2 models were compared by the extra sum-of-squares F test for determining the best model fitting the experimental points. The more complicated model (segmental linear regression) did not significantly (p = 0.4033) fit the experimental points better than the simpler linear regression model. The linear regression model was therefore selected for estimating the infectivity titers in brain preparations of sCJDMM1 patients 2 and 3, and PBS spiked with MF preparation from sCJDMM1 patient 4. Mean incubation periods of animals with positive test results were interpolated to the dose-incubation-period curve of patient 1 to estimate infectivity titers.
The amount of prion infectivity that is below the threshold of detectability of our bioassay but that might be present in the urine of the sCJDMM1 patients was estimated in infectious unit (IU) per milliliter with 95% confidence interval (CI) (rather than in 50% infectious dose [ID50]) by assuming a Poisson distribution in the response variable (32). The total volume of native urine assayed in sCJDMM1 patients 1–3 combined was 84 mL when the 100-fold concentration is taken into account (0.03 mL × 10 mice each in patients 1 and 2 and 0.03 mL × 8 mice in patient 3 × 100-fold concentration). However, considering the 20-fold loss of prion infectivity caused by the use of 100-fold concentrated and dialyzed urine as carrier (see Results), the injected urine equivalents might be 20× lower, i.e., 4.2 (1.5 mL in patients 1 and2, and 1.2 mL in patient 3). Urine infectivity in patient 1 was also estimated by assuming a Poisson distribution based on the volume of raw urine bioassayed from this patient which was 0.99 mL (30 µL × 33 mice).
Histopathologic and Prion Protein Immunohistochemical Analyses
Half brains fixed in formalin and immersed in 98% formic acid for 30 min were sliced into 4 coronal sections and processed for histologic and PrP immunohistochemical analyses (30). Aliquots of BH or sodium phosphotungstate (NaPTA)–precipitated samples, with or without treatment with proteinase K (PK) (specific activity 44 units/mg; Sigma Aldrich, St. Louis, MO, USA) at a concentration of 2 U/mL (45 µg/mL), were loaded onto 15% Tris-glycine sodium dodecylsulfate–polyacrylamide gels, subjected to electrophoresis, and immunoblotted with monoclonal antibody 3F4 (to PrP residues 109–112) (9,33).
Prion Infectivity Titer of Brain Tissue from sCJDMM1
Figure 2
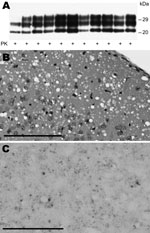
Figure 2. Immunochemical and histopathologic study of humanized transgenic (Tg) mice inoculated with sporadic Creutzfeldt-Jakob disease MM1 (sCJDMM1) microsomal fraction (MF). A) Immunoblot of proteinase K (PK)–resistant scrapie prion protein (PrPSc) from brains...
All mice inoculated with brain MF from the 4 sCJDMM1 patients showed brain histologic lesions, PrPSc deposition patterns, and immunoblot profile of PK-resistant PrPSc observed following sCJDMM1 transmission (Figure 2). The prion infectivity titer in the MF obtained from sCJDMM1 patient 1 was 3.0 × 106 ID50 per gram of tissue equivalent as determined by endpoint dilution bioassay in Tg40 mice (Table 1; Figure 1). We then used incubation times of each mouse inoculated with different brain tissue equivalent dilutions to build up the dose-incubation period curve of Figure 1 (slope ± SE −0.022 ± 0.002; Y-intercept ± SE 10.69 ± 0.73; R2 0.75; p<0.0001). Titers from brain MF of sCJDMM1 patients 2–4 were estimated by interpolating the mean incubation times of the inoculated Tg40 mice to the dose-incubation period curve of Figure 1. The results showed an infectivity titer between 2 and 12 × 106 ID50 per gram of brain tissue equivalent (Table 1). These data indicated that the frontal cortex of all 4 sCJDMM1 patients contained high and comparable prion infectivity titers.
Urine Concentration and Dialysis
To maximize the sensitivity of bioassay, urine samples were concentrated 100× by ultrafiltration and dialyzed against PBS before bioassay in the humanized Tg40 mice. The potential toxic effect of intracerebral injection of concentrated and dialyzed urine was assessed by injecting 3 Tg40 mice with 100× concentrated and dialyzed urine collected from a healthy subject. The inoculated mice showed no clinical signs and were euthanized at 7, 14, and 21 days postinoculation (dpi). The histologic examination revealed no lesions (data not shown). These data show that 100× concentrated and dialyzed urine is not toxic following intracerebral inoculation into Tg40 mice.
Effect of Concentrated and Dialyzed Urine on Prion Infectivity
The effect on prion infectivity of the concentrated and dialyzed urine when used as carrier was assessed by comparing infectivity titers of MF from cerebral cortex of sCJDMM1 patient 4 diluted either in PBS or in 100× concentrated and dialyzed urine. The bioassay analyses showed that using concentrated and dialyzed urine as carrier the infectivity titer of MF was reduced ≈20-fold (1.3 log reduction) (Table 2), indicating that 100× concentrated and dialyzed urine can support prion infectivity although less efficiently than PBS.
Effect of Concentration and Dialysis Procedures on Prion Infectivity
The possible loss of prion infectivity during concentration and dialysis procedures was assessed by bioassay. We compared the infectivity titers of brain MF from sCJDMM1 patient 3 spiked in normal urine before the concentration and dialysis procedure with the infectivity of the same amount of MF spiked in normal urine already concentrated and dialyzed (Table 3). Similar mean incubation periods of 265 days and 268 days, respectively, and prion titers of 8.2 × 106 ID50/g and 6.9 × 106 ID50/g, respectively, were found in the 2 differently processed samples, showing that the concentration and dialysis procedure per se had no detectable effect on infectivity.
Bioassay of Concentrated and Dialyzed Urine and Raw Urine from sCJDMM1 Patients
Figure 3

Figure 3. Immunochemical and histopathologic study of humanized transgenic (Tg) mice inoculated with urine from patients with sporadic Creutzfeldt-Jakob disease MM1 (sCJDMM1). A) Immunoblot of brain homogenates (BH) from Tg40 mice inoculated with...
To detect the prion infectivity in urine from sCJDMM1 patients, urine samples collected from 3 end-stage sCJDMM1 patients were concentrated 100× by ultrafiltration followed by dialysis against PBS. The concentrated and dialyzed urine from the 3 donors was intracerebrally inoculated into 8–10 Tg40 mice. Over the expected normal lifespan (up to 788 dpi), no mice showed any evidence of clinical disease (Table 4). No PK-resistant PrPSc was detected by Western blot, even after enrichment with NaPTA precipitation (Figure 3, panel A). Results of histopathologic and PrP immunohistochemical examinations also were negative (Figure 3, panel B).
On the basis of these negative results, we estimate that prion infectivity in urine of sCJDMM1 patients to be 0 IU/total volume of inoculated urine, which by Poisson distribution is less than 0.37 IU for patients 1 and 2 and 0–0.46 IU for patient 3 (upper limit of 95% CI). However, if we take into account the 100× concentration of urine and the finding that 100× concentrated and dialysized urine may reduce prion infectivity by ≈20-fold (Table 2), the estimates of the highest prion titers possible (i.e., the upper 95% CI) of urine in sCJDMM1 are 0.24 IU/mL for patients 1 and 2 and 0.38 IU/mL for patient 3 (see Materials and Methods).
To directly assess the prion infectivity in urine without concentration and dialysis, we inoculated 33 Tg40 mice with raw urine from sCJDMM1 patient 1. No inoculated mice showed clinical signs of prion disease over their normal lifespan (up to 857 dpi) (Table 4). Similarly, histopathologic and PrP immunohistochemical examination were negative, and no PK-resistant PrPSc was detected by Western blot in the brain even after PrPSc enrichment with NaPTA (data not shown). Thus, the estimated infectivity titer in raw urine of sCJDMM1 by Poisson distribution is 0–0.11 IU/mL (95% CI), not dissimilar to the value (0–0.24) obtained with the same urine after concentration and dialysis.
The present study demonstrates that the urine from patients affected by advanced sCJDMM1, the most common sCJD subtype that alone accounts for ≈60% of all human prion diseases, contains either no prion infectivity or an infectivity titer that is below the detection limit of our bioassays. The bioassays were done in Tg mice expressing human PrP-129M (Tg40) following inoculation with urine obtained from patients with sCJDMM1 and a variety of positive and negative controls. In limit dilution experiments, Tg40 mice inoculated with MF preparations obtained from the brains of 3 urine donors with sCJDMM1 had prion disease develop at up to 105 or 104 dilutions of the brain tissue equivalent depending on whether the MF preparations were inoculated directly or after spiking into concentrated and dialyzed normal human urine.
To enhance the sensitivity of our system, urine samples were concentrated and dialyzed before inoculation. Similar procedures have been used in all the previous studies on prion infectivity of urine (13,22,23), except for the study by Gregori et al. (21). Our procedure is similar to that used by Seeger et al., who reported the detection of prion infectivity in urine from scrapie inoculated mice affected by nephritis (23). Although we demonstrated in the spiking experiment with MF from sCJDMM1 that the 100× concentration and dialysis procedure did not cause infectivity loss (Table 3), the infectivity of the prion-spiked preparation decreased 20-fold when 100× concentrated and dialyzed urine was used as carrier compared with PBS. On the basis of these findings, we estimated that if prion infectivity is present at all in sCJDMM1 urine, it is at most 0.38 IU/mL if the 20-fold infectivity loss is factored in. Because the nature of the potential PrPSc in urine from CJD patients is not known, this urine PrPSc species might show even higher loss of infectivity in the concentrated and dialyzed urine carrier than the brain PrPSc preparations used in the spiking experiments. To address this concern, we inoculated 33 Tg40 mice with raw urine from one of the 3 donors with sCJDMM1. No recipient mice showed evidence of prion disease suggesting an infectivity ranging from 0.0 and 0.11 IU/per mL (upper limit of 95% CI) as estimated by the Poisson distribution.
Although asymptomatic disease in recipient mice associated with NaPTA- undetectable minute amounts of PrPSc cannot be excluded, our inability to detect prion infectivity in human urine of patients with sCJDMM1 differs from several recent experimental studies on urine of prion-affected animals. Low prion infectivity has been demonstrated in urine from scrapie-infected hamsters (21,22), CWD-infected deer (13), and in scrapie-infected mice affected by lymphocytic nephritis. In the last study, however, no urine infectivity was found in non-nephritic mice (23). Three additional studies have demonstrated the presence of PrPSc in urine from scrapie-infected hamsters and CWD-infected deer using protein misfolding cyclic amplification (PMCA) (13,34,35). However, this highly sensitive procedure can detect prion concentrations below the level of detectability of bioassays.
The most likely explanation for the discrepancy between our negative results on human urine and the positive findings by bioassay in urine from animals resides in the different locale and mode of formation of the prion agents. In all the published animal experiments, including bioassays and PMCA, the prion disease was induced by intracerebral or oral administration of exogenous prions, whereas we examined urine infectivity in a naturally occurring sporadic human prion disease. In exogenously acquired prion diseases, PrPSc is much more likely to be widely present in nonneural peripheral organs, including blood, kidney, and bladder, which is likely the result of early exposure of peripheral organs to the inoculated prions. In sCJD, which is believed to occur spontaneously in the brain (rather than being acquired by infection from exogenous prions), only minute amounts of PrPSc have been detected in a few nonneural peripheral organs and tissues such as skeletal muscle and spleen (6–8,10). In contrast, in variant CJD (vCJD), the form of CJD acquired by consumption of BSE-infected beef, the spread of PrPSc to peripheral organs is much wider and typically involves lymph nodes, tonsil, spleen, portions of the intestinal tract, and the skeletal muscle (7,10,36,37), as well as kidney and other organs (9). These considerations indicate that vCJD (not sCJD) in principle is more similar to the exogenously acquired animal prion diseases that have been used to study prion infectivity in urine. Therefore, vCJD urine that is more likely to contain prion infectivity should be tested by PMCA or bioassay. However, the reported infectivity of animal urine might also result, at least in some instances, from contamination with feces.
Recent data have proved that feces from hamsters infected with scrapie by the oral route and to a lesser extent through intracerebral and intraperitoneal inoculation, contain a discrete amount of PrPSc and prion infectivity (38,39). Infectivity has been demonstrated also in feces from deer orally infected by CWD (40). In hamsters and mice, metabolic cages were used for urine collection, a method in which cross-contamination by feces may actually occur. However, prion infectivity was also demonstrated in urine from CWD-infected deer from which urine collection could easily be performed by catheterization (although this procedure was not mentioned by the authors) (13).
Although additional studies are still needed to determine whether minute amounts of prion infectivity or PrPSc are present in urine from patients with sCJD and to assess the presence of infectious prion in urine from patients with every other form and subtype of human prion diseases, our study shows that urine from patients with sCJDMM1, the most common subtype of sCJD, does not contain prion infectivity detectable by our bioassay and suggests that no significant prionuria occurs in this common subtype of human prion disease.
Dr Notari is an instructor at the Department of Pathology, Case Western Reserve University, Cleveland, Ohio, USA. His research interests are the prion diseases with particular focus on the characteristics of PrPSc in different CJD subtypes and their relationships with infectivity.
Acknowledgments
We thank Diane Kofskey for providing histologic and immunochemical preparations; Fusong Chen, Michael Payne, and Meiling Wang for caring for the animals; and Qiwei Yang and Yvonne Cohen for assistance with urine preparations.
This study was supported by an award from Ferring Pharmaceuticals A/S and Instituto Massone, and by National Institutes of Health grants AG014359 and NS052319, grant UR8/CCU515004 from the Centers for Disease Control and Prevention, and the Charles S. Britton Fund.
References
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–39. DOIPubMedGoogle Scholar
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. DOIPubMedGoogle Scholar
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–7. DOIPubMedGoogle Scholar
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–5. DOIPubMedGoogle Scholar
- Glatzel M, Abela E, Maissen M, Aguzzi A. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N Engl J Med. 2003;349:1812–20. DOIPubMedGoogle Scholar
- Peden AH, Ritchie DL, Head MW, Ironside JW. Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:927–35. DOIPubMedGoogle Scholar
- Beekes M, McBride PA. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J. 2007;274:588–605. DOIPubMedGoogle Scholar
- Notari S, Moleres FJ, Hunter SB, Belay ED, Schonberger LB, Cali I, Multiorgan detection and characterization of protease-resistant prion protein in a case of variant CJD examined in the United States. PLoS ONE. 2010;5:e8765. DOIPubMedGoogle Scholar
- Brown P, Andreoletti O, Bradley R, Budka H, Deslys JP, Groschup M, WHO tables on tissue infectivity distribution in transmissible spongiform encephalopathies. World Health Organization. 2010 [cited 2011 Nov 2]. http://www.who.int/bloodproducts/tablestissueinfectivity.pdf
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV. Warren RJ, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314:133–6. DOIPubMedGoogle Scholar
- Mathiason CK, Hays SA, Powers J, Hayes-Klug J, Langenberg J, Dahmes SJ, Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS ONE. 2009;4:e5916. DOIPubMedGoogle Scholar
- Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS ONE. 2009;4:e4848. DOIPubMedGoogle Scholar
- Maddison BC, Rees HC, Baker CA, Taema M, Bellworthy SJ, Thorne L, Prions are secreted into the oral cavity in sheep with preclinical scrapie. J Infect Dis. 2010;201:1672–6. DOIPubMedGoogle Scholar
- Lacroux C, Simon S, Benestad SL, Maillet S, Mathey J, Lugan S, Prions in milk from ewes incubating natural scrapie. PLoS Pathog. 2008;4:e1000238. DOIPubMedGoogle Scholar
- Ligios C, Cancedda MG, Carta A, Santucciu C, Maestrale C, Demontis F, Sheep with scrapie and mastitis transmit infectious prion through the milk. J Virol. 2011;85:1136–9. DOIPubMedGoogle Scholar
- Shaked GM, Shaked Y, Kariv-Inbal Z, Halimi M, Avraham I, Gabizon R. A protease-resistant prion protein isoform is present in urine of animals and humans affected with prion diseases. J Biol Chem. 2001;276:31479–82. DOIPubMedGoogle Scholar
- Furukawa H, Doh-ura K, Okuwaki R, Shirabe S, Yamamoto K, Udono H, A pitfall in diagnosis of human prion diseases using detection of protease-resistant prion protein in urine. Contamination with bacterial outer membrane proteins. J Biol Chem. 2004;279:23661–7. DOIPubMedGoogle Scholar
- Serban A, Legname G, Hansen K, Kovaleva N, Prusiner SB. Immunoglobulins in urine of hamsters with scrapie. J Biol Chem. 2004;279:48817–20. DOIPubMedGoogle Scholar
- Head MW, Kouverianou E, Taylor L, Green A, Knight R. Evaluation of urinary PrPSc as a diagnostic test for sporadic, variant, and familial CJD. Neurology. 2005;64:1794–6. DOIPubMedGoogle Scholar
- Gregori L, Kovacs GG, Alexeeva I, Budka H, Rohwer RG. Excretion of transmissible spongiform encephalopathy infectivity in urine. Emerg Infect Dis. 2008;14:1406–12. DOIPubMedGoogle Scholar
- Kariv-Inbal Z, Ben-Hur T, Grigoriadis NC, Engelstein R, Gabizon R. Urine from scrapie-infected hamsters comprises low levels of prion infectivity. Neurodegener Dis. 2006;3:123–8. DOIPubMedGoogle Scholar
- Seeger H, Heikenwalder M, Zeller N, Kranich J, Schwarz P, Gaspert A, Coincident scrapie infection and nephritis lead to urinary prion excretion. Science. 2005;310:324–6. DOIPubMedGoogle Scholar
- Dagdanova A, Ilchenko S, Notari S, Yang Q, Obrenovich ME, Hatcher K, Characterization of the prion protein in human urine. J Biol Chem. 2010;285:30489–95. DOIPubMedGoogle Scholar
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–80. DOIPubMedGoogle Scholar
- Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci U S A. 1996;93:15457–62. DOIPubMedGoogle Scholar
- Van Dorsselaer A, Carapito C, Delalande F, Schaeffer-Reiss C, Thierse D, Diemer H, Detection of prion protein in urine-derived injectable fertility products by a targeted proteomic approach. PLoS ONE. 2011;6:e17815. DOIPubMedGoogle Scholar
- Tateishi J, Sato Y, Koga M, Doi H, Ohta M. Experimental transmission of human subacute spongiform encephalopathy to small rodents. I. Clinical and histological observations. Acta Neuropathol. 1980;51:127–34. DOIPubMedGoogle Scholar
- Kuroda Y, Gibbs CJ Jr, Amyx HL, Gajdusek DC. Creutzfeldt-Jakob disease in mice: persistent viremia and preferential replication of virus in low-density lymphocytes. Infect Immun. 1983;41:154–61.PubMedGoogle Scholar
- Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25:7944–9. DOIPubMedGoogle Scholar
- Reichl H, Balen A, Jansen CA. Prion transmission in blood and urine: what are the implications for recombinant and urinary-derived gonadotrophins? Hum Reprod. 2002;17:2501–8. DOIPubMedGoogle Scholar
- Gregori L, McCombie N, Palmer D, Birch P, Sowemimo-Coker SO, Giulivi A, Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet. 2004;364:529–31. DOIPubMedGoogle Scholar
- Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J, Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem. 2008;283:30557–65. DOIPubMedGoogle Scholar
- Murayama Y, Yoshioka M, Okada H, Takata M, Yokoyama T, Mohri S. Urinary excretion and blood level of prions in scrapie-infected hamsters. J Gen Virol. 2007;88:2890–8. DOIPubMedGoogle Scholar
- Gonzalez-Romero D, Barria MA, Leon P, Morales R, Soto C. Detection of infectious prions in urine. FEBS Lett. 2008;582:3161–6. DOIPubMedGoogle Scholar
- Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet. 2001;358:171–80. DOIPubMedGoogle Scholar
- Hilton DA, Sutak J, Smith ME, Penney M, Conyers L, Edwards P, Specificity of lymphoreticular accumulation of prion protein for variant Creutzfeldt-Jakob disease. J Clin Pathol. 2004;57:300–2. DOIPubMedGoogle Scholar
- Safar JG, Lessard P, Tamgüney G, Freyman Y, Deering C, Letessier F, Transmission and detection of prions in feces. J Infect Dis. 2008;198:81–9. DOIPubMedGoogle Scholar
- Krüger D, Thomzig A, Lenz G, Kampf K, McBride P, Beekes M. Fecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Vet Res. 2009;40:4. DOIPubMedGoogle Scholar
- Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Asymptomatic deer excrete infectious prions in feces. Nature. 2009;461:529–32. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 18, Number 1—January 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Pierluigi Gambetti or Qingzhong Kong, Department of Pathology, 2085 Adelbert Road, Cleveland, OH 44106, USA
Top