Volume 28, Number 6—June 2022
Research
Geographic Origin and Vertical Transmission of Leishmania infantum Parasites in Hunting Hounds, United States
Cite This Article
Citation for Media
Abstract
Vertical transmission of leishmaniasis is common but is difficult to study against the background of pervasive vector transmission. We present genomic data from dogs in the United States infected with Leishmania infantum parasites; these infections have persisted in the apparent absence of vector transmission. We demonstrate that these parasites were introduced from the Old World separately and more recently than L. infantum from South America. The parasite population shows unusual genetics consistent with a lack of meiosis: a high level of heterozygous sites shared across all isolates and no decrease in linkage with genomic distance between variants. Our data confirm that this parasite population has been evolving with little or no sexual reproduction. This demonstration of vertical transmission has profound implications for the population genetics of Leishmania parasites. When investigating transmission in complex natural settings, considering vertical transmission alongside vector transmission is vital.
Leishmaniasis is a disease caused by obligate intracellular protozoan parasites of the genus Leishmania, including Leishmania infantum (1). Zoonotic visceral leishmaniasis (ZVL) occurs in countries to which the disease is endemic and enzootic in human and animal populations. Dogs are the predominant domestic reservoir of ZVL and thus play a critical role in its ecology and control. Seropositivity is often evident in dogs before visceral leishmaniasis (VL) can be observed in humans (2), and dog ownership is a risk factor for human disease (3–5). As such, control measures in locations where ZVL is prominent include insecticide treatment or culling of dogs.
Although ZVL is transmitted primarily through phlebotomine sand flies (6), the role of other means of transmission, particularly vertical transmission, has been demonstrated (7–10). Transplacental transmission of L. infantum parasites can maintain infection within dog populations (8,9); pups have been shown to be infected in utero (11–13). Vertical transmission is not unique to dogs (14,15), and case reports have identified vertical transmission of VL as a cause of infant illness and death in humans (16,17). Beyond these reports, little is known about the risks of vertical transmission in dogs or humans. Leishmania parasites are thought to replicate exclusively clonally as intracellular amastigotes in vertebrate hosts. In contrast, in sand flies they undergo transformation into promastigotes, where they can still reproduce clonally but can also undergo meiosis to complete sexual reproduction (18,19), although sexual reproduction is not obligatory for transmission. Nothing is known about the transmission genetics of vertically transmitted Leishmania populations (8,20,21) or how the absence of vector stages affects the establishment or pathogenicity of mammalian infections.
In the United States, leishmaniasis is enzootic in hunting dogs. ZVL was first identified in 1980 in a dog with no travel outside of the United States. A large outbreak in 1999 prompted an investigation by the Centers for Disease Control and Prevention to determine the burden of disease in US hunting hounds (22,23). This investigation established the likely introduction of infected dogs from ZVL-endemic areas of Europe through the United Kingdom, but no testing of dogs outside the United States was performed, and genomic similarity to L. infantum parasites from Europe and South America was not evaluated (23,24).
We subsequently established the primary route of transmission as vertical from dam to pup (9,25). Despite extensive surveillance associated with these infected dogs (26,27), no naturally L. infantum–infected sand fly has been found in the United States. Although vector transmission of L. infantum parasites from these hunting dogs has been experimentally demonstrated (27,28), it does not appear to be involved in these natural infections.
We examined whole-genome sequences of L. infantum parasites from canine autochthonous infection within the United States and sought to identify a likely geographic origin. We looked for evidence of recombination between these L. infantum isolates to test for genomic evidence of predominantly vertical transmission. Many dogs are imported from ZVL-endemic areas to non–ZVL-endemic areas; our findings highlight the need for increasing awareness and testing before import of dogs from ZVL-endemic countries (29).
Ethics
All dogs were enrolled with informed consent from their caretakers, and protocols followed were approved by the University of Iowa Institutional Animal Care and Use Committee. This AAALAC International–accredited institution follows the requirements for the US National Institutes of Health Office of Laboratory Animal Welfare Assurances and operates under the 2015 reprint of the Public Health service Policy on Humane Care and Use of Laboratory Animals.
Sample Collection of Parasites from US Hunting Dogs
The 7 L. infantum samples from US hunting dogs used in this study were identified during a retrospective cohort study of L. infantum infection in US hunting dogs (26,27,30). To identify Leishmania-infected dogs, an active surveillance cohort of 4 large (>50 dogs each) kennels was established from 3 different states in the midwestern United States during 2007–2017. Licensed veterinarians collected 1–5 mL whole blood and serum samples from all dogs at these kennels. Dogs were considered infected if they were positive by quantitative PCR detecting Leishmania-specific DNA and had Leishmania-specific antibodies (31). Parasites from the buffy coat of Leishmania-positive dogs were cultured in both Schneider and HOMEM media overnight at 26°C then placed onto agar slants and incubated for 3–4 weeks and observed daily for growth. Parasite cultures include 1 sibling pair (foxymo_01, foxymo_02); remaining dogs all have different grandparents. Because of the frequent exchange of hunting dogs among kennels and states, within 2 generations the ancestors of the sampled dogs came from 12 kennels and 9 different US states (Georgia, Illinois, Iowa, Kansas, Minnesota, Missouri, New Jersey, New York, and Virginia) that included the primary US locations for hunting hound breeding.
Whole-Genome Sequencing of Parasite DNA from Hunting Dogs
We used QIAamp DNA Blood Mini Kit (QIAGEN, https://www.giagen.com) according to manufacturer specifications to isolate DNA directly from primary parasite cultures. We thawed parasite cultures, counted, and placed 1 million parasites into Trizol Reagent (ThermoFisher Scientific, https://www.thermofisher.com) and extracted according to manufacturer specifications. We assessed quality and quantity of isolated DNA by using NanoDrop 2000 (ThermoFisher Scientific).
DNA Sequencing
We sheared DNA into 400–600-bp fragments by focused ultrasonication using the Covaris Adaptive Focused Acoustics technology (Covaris, https://www.covaris.com). We performed 2 methods of DNA sequencing, depending on the amount of DNA supplied, by using the NEBNext DNA Library Prep kit (New England BioLabs, https://www.neb.com). For volumes <500 ng, we amplified libraries by using KAPA HiFI DNA polymerase (Kapa Biosystems, https://kapabiosystems.com) and generated 100-bp paired-end reads on the Illumina HiSeq 2000 (Illumina, https://www.illumina.com). For volumes >500 ng, we generated amplification-free libraries and obtained 150-bp paired-end reads on the Illumina HiSeq X10 (Illumina). We performed sequencing following manufacturers’ standard protocols.
Genomic Analysis Pipeline
We analyzed the genomic data of 7 L. infantum US hound isolates with an additional 92 publicly available L. infantum isolates sampled from a global distribution (Appendix 1). For all samples, we subjected newly generated and downloaded fastq files to identical analysis pipelines. We trimmed reads using Trimmomatic version 0.39 (http://www.usadellab.org/cms/?page=trimmomatic) (parameters “ILLUMINACLIP:PE_adaptors.fa:2:30:10 TRAILING:15 SLIDINGWINDOW:4:15 MINLEN:50”) and mapped them against the reference genome of JPCM5 v45 (https://tritrypdb.org) with BWA version 0.7.17 (bwa mem -M option) (32). Single-nucleotide polymorphisms (SNPs) were called using GATK version 4.1.2.0 (33): HaplotypeCaller was used with parameters “-ERC GVCF–annotate-with-num-discovered-alleles–sample-ploidy 2” to generate gvcf files for each sample, then combined using “GenomicsDBImport” and genotyped with “GenotypeGVCFs.” Calls were filtered with “VariantFiltration” (filters: “QD<2.0, MQ<50.0, FS>20.0, SOR>2.5, BaseQRankSum<-3.1, ClippingRankSum<-3.1, MQRankSum<-3.1, ReadPosRankSum<-3.1and DP<6”) and only polymorphic SNPs retained. We removed SNPs with >20% missing calls across samples, reducing the total number of SNPs from 43,528 to 43,336.
Phylogenetic Reconstruction and Admixture Analysis
We performed phylogenetic reconstruction by using distance-based and maximum-likelihood methods on genome-wide genotype calls. For the distance-based approach, we calculated pairwise Nei D distances and reconstructed trees by the neighbor-joining method using the R packages StAMPP version 1.6.1 (34) and ape version 5.4. We based bootstrap values on 100 replicates. For maximum-likelihood phylogenies, we converted the vcf file to fasta format with IUPAC codes using bcftools consensus. We estimated 1,000-bootstrap maximum-likelihood phylogenies by using RAxML-NG version 0.8.1-c1 (35) and the GTJC model that captures changes between heterozygous and homozygous states.
We preprocessed genome-wide SNPs for admixture analysis version 1.3.0 (36) only with plink version 1.90 changing the vcf format into ped and map format and removing SNPs with a missing fraction of >0.05 and variants closer to each other than 2,000-bp with the arguments “–geno” and “–bp-space.” We ran admixture for values of K from 1 to 20 and optimal numbers of groups (K) were chosen on the basis of lowest cross-validation error (Appendix 2 Figure 1). Because there was no clear number of K at which the cross-validation error plateaued, we present analyses with the smallest K at first sign of plateauing of the error and 2 larger Ks with smaller errors.
Molecular Clock Dating
We used 2 molecular clock approaches. The first method was a simple clock model using PATHd8 (37) for all RAxML-NG bootstrap trees, constraining the root of the non-US New World clade to 537 years ago. The second method was a Bayesian approach that used BEAST version 1.10.4 (https://beast.community) to enable flexible modeling of rate variation with standard substitution models, a narrow uniform prior of 536.9–537.1 years for the New World clade and leaf heights set to the year of collection (Appendix 1), or constrained to 2005–2007 for samples from (39) and to 1900–2020 for the sample ‘DOG_STRAIN’ of unknown sampling date (38). New World and US hound clades were constrained to be monophyletic, and Bayesian Markov Chain Monte Carlo analysis was initialized with the RAxML-NG phylogeny for concatenated chromosomes. The substitution model was Hasegawa-Kishino-Yano with a 4-category gamma distribution of rate variation across sites. Results are based on 8 independent Bayesian Markov Chain Monte Carlo chains of 10 million generations, 1 million generations burn-in, and convergence checked using Tracer version 1.7.1 (https://beast.community/tracer). We accepted analyses if 6 out of 8 chains were at similar likelihoods for 2 million generations. Remaining parameters were defaults from Beauti version 1.10.4. Only results for both strict and uncorrelated gamma-distributed clocks converged and are shown.
Population Genomics Analysis
We grouped parasite samples according to geographic origin and isolated host type (Table). Groups were characterized by their number of segregating SNPs, inbreeding coefficients, and linkage decay with distance. We performed analysis in R (R Foundation for Statistical Computing, https://www.r-project.org) with the exception of R2 estimates, which we estimated as genotype correlations with vcftools version 0.1.16 (41) and parameters “–geno-r2” and “–interchrom-geno-r2.” We used genotype correlations because haplotypes cannot be accurately phased for our small population sets. We calculated the inbreeding coefficient F based on the formula F = 1 –((cAB/N)/(2 × fA × fB)), where cAB represents the heterozygote count, N the group size, and fA and fB the frequency of alleles A and B.
Aneuploidy Estimation
We estimated sequencing coverage on the basis of sample-specific mapped bam files. For each sample, indels were determined and indel realignment was performed with the GATK version 3.6 (33) tools “RealignerTargetCreator” and “IndelRealigner.” Quality filtering and duplicate removal was done with samtools version 1.3 using the parameters “-F 1024 -f 0x0002 -F 0x0004 -F 0x0008.” Coverage was estimated with bedtools version 2.17.0 (42) genomecov and parameters “-d -split.” For each sample, the median coverage per chromosome was assumed to represent the diploid state, so chromosome somy = (chromosome_coverage/median_coverage) × 2. Allele frequencies for isolate-specific SNPs were estimated on the basis of previous bam files and quality filtered with samtools “-q 20 -f 0x0002 -F 0x0004 -F 0x0008.” Coverage by genomic position was obtained with samtools mpileup “-d 3500 -B -Q 10” and transformed into sync format with mpileup2sync “–min-qual 20” (43).
Independent Introduction of US Hound–Derived Parasites from the Mediterranean Region
To assess the geographic origins of L. infantum parasites within US hunting dogs, we generated whole-genome sequence data for 7 L. infantum isolates from outbred hounds from 4 kennels in the midwestern United States and an ancestry tracing back to kennels in 9 US states within 2 generations with haploid coverage ranging from 29 to 78 (median 69). We compared these samples with 92 previously published L. infantum genome sequences of other strains from other global populations (38,39,44) (Appendix 1).
Figure 1
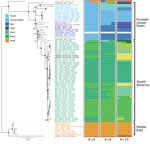
Figure 1. Neighbor-joining tree based on pairwise Nei distances demonstrating geographic origin of US hound Leishmaniaisolates. Phylogenies were reconstructed on the basis of whole-genome genotype calls of 83 parasite samples...
Figure 2
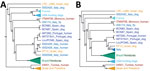
We constructed distance-based and ML phylogenies from whole-genome SNP variants to compare L. infantum genomes from US dogs to samples from L. infantum–endemic regions of South America and the Old World. Parasites from US hounds were monophyletic, part of the L. infantum MON-1 clade (38,45), and clearly distinct from L. infantum isolates from South America (Figure 1; Appendix 2 Figure 2). These factors suggest independent introduction to the New World. The genetically closest parasite samples were from southern Europe, but the exact origin was ambiguous. Distance-based methods suggested 4 samples from France as genetically most closely related to US isolates (Figure 1; Figure 2, panel A). The ML phylogeny placed US parasites close to a more widespread group of MON-1 parasites (Figure 2, panel B).
To further investigate parasites’ relatedness, we performed admixture analysis, which was consistent with the phylogenetic results. We applied cross validation, a standard approach in admixture to determine an optimal number of populations (K) that best explains the relatedness between samples. Because this process did not identify a single optimal K (Appendix 2 Figure 1), we considered more than one K (Figure 1; Appendix 2 Figure 2). We concentrated our analysis on 83 core samples consisting of samples from the United States and other samples from the MON-1 clade (Figures 1, 2). For K = 4 populations, US hound parasites were placed together with all remaining samples from Europe and single samples from Israel and Morocco (Figure 1). For K = 6 and K = 15, US samples formed a separate group, only inferred to share ancestry with one sample from Italy and one from Morocco for K = 6. A similar pattern was present within the total set of 99 samples. For K = 7, US and 2 parasites from France grouped together, and for K = 11, US samples only shared substantial variation with 1 sample from Italy (Appendix 2 Figure 2, panel A), which together suggested a clear origin from Mediterranean Europe but no clear country of origin.
Molecular Clock Dating Confirms Recent Divergence of US Hound–Derived Parasites
Figure 3
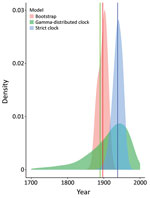
Figure 3. Molecular clock estimates of the date of the most recent common ancestor of US hound Leishmaniasamples. Shaded densities are normal kernel densities for the bootstrap estimates from PATHd8...
We dated the independent introduction of US hound parasites by using 2 different molecular clock approaches, relying on previously estimated introduction of L. infantum parasites into the New World ≈500 years ago (46). The first analysis using our maximum-likelihood phylogeny estimated the mean date of divergence between US parasites and relatives from Europe as 1897 (95% CI 1873–1917), whereas 2 Bayesian approaches produced estimates of 1938 (strict clock, 95% highest posterior density CI 1910–1965) and 1889 (relaxed clock, 95% CI 1689–1991) (Figure 3). Estimates across a range of approaches thus suggest that US hound parasites were introduced much more recently than L. infantum parasites were introduced to South America.
Patterns of Heterozygosity in US Hound Parasites Suggest Clonal Evolution
Figure 4
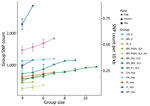
Figure 4. Number and density of segregating SNPs in each group of Leishmania infantumisolates by geographic region and type of host. Values are shown as both the number (left y-axis)...
The genetic variation in a population should reflect its reproductive biology. We thus compared variation in US hound parasites with L. infantum populations isolated from dogs in areas where vector transmission occurs and with populations isolated from humans or a mixture of both hosts in other parts of the world (Table). Within-population diversity of the US hound parasites was intermediate between the high diversity of populations from the Old World and the low diversity of parasites from different regions within Brazil (Figure 4). For most populations, the number of polymorphic sites increased with sample size, indicating that increasing numbers of rare variants were detected with larger sample sizes. This sample size–based increase was minimal in the US hound parasite population, suggesting a large proportion of shared variation among these isolates.
Figure 5

Figure 5. Extreme excess of heterozygous sites in the US hound–derived Leishmania infantumisolates. The group-specific inbreeding coefficient F is shown for all polymorphic sites in the respective parasite population. F...
To explore this shared variation further, we directly estimated population heterozygosity through the inbreeding coefficient F and the fraction of population-specific polymorphic heterozygous SNP sites (Figure 5; Appendix 2 Figure 3). The inbreeding coefficient was significantly different between populations (Kruskal-Wallis test, χ2 = 2843.1, df = 12; p<0.001), and the US hound parasite population had exceptionally low F values compared with all other populations (Dunn test, adjusted; p<0.001) (Figure 5). This difference was largely caused by 79% of all polymorphic sites within US hound–derived parasites sharing the same heterozygous genotype across all 7 sampled hound isolates. This extreme excess of shared heterozygosity is present across all chromosomes and is in strong contrast to the remaining populations. Absolute numbers of heterozygous sites in the US samples were higher than in other populations (Table; Appendix 2 Figure 4, panel A). This difference could be caused by either the accumulation of mutations during a period of clonal evolution shared by these samples or a hybrid origin of the founder strain of our US samples between 2 closely related L. infantum populations (Appendix 2 Figure 4, panel B), because clonal propagation would maintain any heterozygosity.
No Evidence for Sexual Reproduction in L. infantum Isolated from US Hounds
Figure 6

Figure 6. Decay of linkage disequilibrium with genomic distance across geographically confined groups of Leishmania infantumisolates. A) US_d_S5, B) BR_d_A5, C) IS_d_A5, D) BR_RGN_VLh_A5, E) BR_RGN_VLh_Ah_S5, F) BR_MA_VLh_Ah_S5, G) BR_MG_VLh_S5....
If L. infantum parasite transmission in US hunting dogs occurs solely through vertical transmission, we would expect genomic signatures of sexual reproduction to be absent because sexual reproduction is thought to be limited to the vector stage (18). Sexual reproduction returns proportions of heterozygous and homozygous variants to the Hardy-Weinberg equilibrium. We propose that the observed extreme excess of shared heterozygous sites in US hound parasites is possible because these parasites evolve clonally for many generations with no mechanism to reduce the number of heterozygous sites through sexual reproduction. To test this proposition, we investigated whether genetic linkage between pairs of SNPs reduces as the distance between loci increases, which would be expected if recombination is occurring. Almost all global L. infantum populations showed this expected decay in linkage within chromosomes, except US hound–derived parasites and 2 populations from Brazil (Figure 6). The 2 populations from Brazil had too few polymorphic sites to reliably assess linkage patterns. The US hound parasites also had relatively few sites for analysis, because unphased shared heterozygous sites cannot be used for linkage estimation. However, the remaining loci showed no evidence of linkage decay with genetic distance. Pairs of variants on different chromosomes showed very similar linkage to within-chromosome comparisons (Figure 6). This finding indicates that evidence for meiotic recombination in the US dog L. infantum population is lacking.
Reduced Variation in Aneuploidy in Mammalian Host–Derived Parasites
Figure 7

Figure 7. Aneuploidy variation of Leishmaniaisolates from US hunting hounds. A) Aneuploidy profiles, shown as a heatmap of estimated somy for each isolate and chromosome. The sample phylogeny is extracted...
Leishmania populations frequently show variation in copy number of individual chromosomes with frequent aneuploidy turnover even within a clonal population (mosaic aneuploidy). Aneuploidy variation between US isolates was largely limited to one third of the chromosomes and variation did not correlate to chromosome-specific heterozygosity, which should have been reduced if aneuploidy turnover was high (Figure 7; Appendix 2 Figure 5). Although this estimate of aneuploidy variation through mean ploidy profiles between isolates is conservative, it supports initial findings that aneuploidy turnover might be greater in cultured promastigotes versus intra-host amastigotes (47,48).
Our data confirm that L. infantum found in US hounds represents an independent introduction of Leishmania into the New World. Although we cannot be definitive about the precise origin of US hound L. infantum isolates, they form part of the MON-1 clade, associated with canine leishmaniasis throughout the Mediterranean region. Closely related MON-1 samples are from Mediterranean Europe, consistent with epidemiologic findings that deer hunting hounds imported from France may have introduced L. infantum parasites into the US hound population, potentially through UK breeding connections (29).
Molecular clock analyses suggested that US hound parasites diverged from other L. infantum isolates around 1900, but parasitized dogs could have entered the United States more recently. These date estimates also depend on the assumed origin of the main New World subspecies (L. infantum subspecies chagasi) 537 years ago, the central estimate from an analysis of microsatellite data, although with very wide CIs (46). The safest interpretation of our analysis is therefore a much more recent divergence of US canine parasites from parasites in Europe than the main New World clade of L. infantum subsp. chagasi.
Our data confirmed the highly unusual genetics of the L. infantum population in US hounds. This parasite population demonstrated an excess of shared heterozygous loci, which could have been initiated by an already heterozygous founder strain. However, the preservation of heterozygous sites across our US samples is consistent with clonal reproduction, which is also confirmed by the absence of any signature of reduction in genetic linkage with genomic distance in this population. Without a broader sampling of parasites from US hounds, we cannot rule out that transmission via sand flies is occurring elsewhere in the United States. Similarly, we cannot quantify the amount of parasite sexual reproduction from these data and so cannot completely rule out that sexual reproduction and therefore vector transmission are occurring. However, our results are consistent with parasites replicating only clonally as amastigotes in dog phagocytes in the absence of sand fly vectors. No sand fly transmission of L. infantum parasites from dogs in the United States has been demonstrated (7,9,28), so we suspect that transmission within this population is largely occurring vertically and directly between dogs.
The population genetic signatures of vertical transmission we have found could be useful in characterizing the epidemiology of other Leishmania populations. The extent to which these signatures occur in more complex situations, such as with multiple introductions of parasites or mixed vertical and horizontal transmission, remains to be established. The most direct evidence of vertical transmission would be to find that the relatedness between parasite isolates directly reflected the pedigrees of the sampled dogs, although this would be potentially complicated by horizontal transmission between dogs (e.g., through blood-blood contact during fights) (49). Although we have not attempted to test this possibility, parasites from the pair of siblings included here (foxymo_01 and foxymo_02) were genetically closest to each other and clearly separated from all others.
In conclusion, our data confirm the 1999–2000 outbreak investigation finding by the Centers for Disease Control and Prevention that at least 1 L. infantum population in US dogs was a recent introduction from Europe, distinct and much more recent than the main population of L. infantum in South America. This population has reproduced largely or exclusively clonally, presumably as amastigotes within canine hosts. We see no evidence of recent recombination associated with vector transmission up to the limits of our detection levels; thus, transmission has likely occurred either vertically through maternal-offspring transplacental transmission or horizontally through blood-blood contact. The absence of evidence for vector-based transmission in the northern United States makes this an unusual, and perhaps unique, ecologic system. Our findings enable the study of many aspects of Leishmania biology without the complication of occasional vector transmission, including adaptation of parasites to the mammal host without the additional selection pressure of vector transmissibility, mutation rates, and rates of amastigote cell division.
Dr. Franssen is an assistant professor in the division of evolutionary biology at the Ludwig-Maximilians-Universität in Munich, Germany. A bioinformatician by training, she has worked in evolutionary genomics and population genetics throughout her career studying evolution in a diverse range of organisms but since her postdoctorate work at the Wellcome Sanger Institute has focused on Leishmania and leishmaniasis.
Acknowledgments
We thank the dog caretakers who helped us gather this data and previous members of the Petersen laboratory, who collected the samples, particularly Carolyne Bennett and staff veterinarians, who performed physical examinations, ran diagnostics, performed data entry, and ran analyses over the years. We also thank members of the Wellcome Sanger Institute DNA pipelines teams for producing and sequencing DNA libraries.
This research was funded by Wellcome (grant 206194) and the National Institutes of Health (grant R01TW010500).
References
- Miró G, Petersen C, Cardoso L, Bourdeau P, Baneth G, Solano-Gallego L, et al. Novel areas for prevention and control of canine leishmaniosis [Erratum in: Trends Parasitol. 2017;33:718–30]. Trends Parasitol. 2017;33:718–30. DOIPubMedGoogle Scholar
- Lima ID, Lima ALM, Mendes-Aguiar CO, Coutinho JFV, Wilson ME, Pearson RD, et al. Changing demographics of visceral leishmaniasis in northeast Brazil: Lessons for the future. PLoS Negl Trop Dis. 2018;12:
e0006164 . DOIPubMedGoogle Scholar - Gavgani ASM, Mohite H, Edrissian GH, Mohebali M, Davies CR. Domestic dog ownership in Iran is a risk factor for human infection with Leishmania infantum. Am J Trop Med Hyg. 2002;67:511–5. DOIPubMedGoogle Scholar
- Bsrat A, Berhe M, Gadissa E, Taddele H, Tekle Y, Hagos Y, et al. Serological investigation of visceral Leishmania infection in human and its associated risk factors in Welkait District, Western Tigray, Ethiopia. Parasite Epidemiol Control. 2018;3:13–20. DOIPubMedGoogle Scholar
- Lima ÁLM, de Lima ID, Coutinho JFV, de Sousa ÚPST, Rodrigues MAG, Wilson ME, et al. Changing epidemiology of visceral leishmaniasis in northeastern Brazil: a 25-year follow-up of an urban outbreak. Trans R Soc Trop Med Hyg. 2017;111:440–7. DOIPubMedGoogle Scholar
- Bates PA. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol. 2007;37:1097–106. DOIPubMedGoogle Scholar
- Toepp AJ, Bennett C, Scott B, Senesac R, Oleson JJ, Petersen CA. Maternal Leishmania infantum infection status has significant impact on leishmaniasis in offspring. PLoS Negl Trop Dis. 2019;13:
e0007058 . DOIPubMedGoogle Scholar - Grinnage-Pulley T, Scott B, Petersen CA. A mother’s gift: congenital transmission of Trypanosoma and Leishmania species. PLoS Pathog. 2016;12:
e1005302 . DOIPubMedGoogle Scholar - Boggiatto PM, Gibson-Corley KN, Metz K, Gallup JM, Hostetter JM, Mullin K, et al. Transplacental transmission of Leishmania infantum as a means for continued disease incidence in North America. PLoS Negl Trop Dis. 2011;5:
e1019 . DOIPubMedGoogle Scholar - Petersen CA. New means of canine leishmaniasis transmission in north america: the possibility of transmission to humans still unknown. Interdiscip Perspect Infect Dis. 2009;2009:
802712 . DOIPubMedGoogle Scholar - da Silva SM, Ribeiro VM, Ribeiro RR, Tafuri WL, Melo MN, Michalick MSM. First report of vertical transmission of Leishmania (Leishmania) infantum in a naturally infected bitch from Brazil. Vet Parasitol. 2009;166:159–62. DOIPubMedGoogle Scholar
- Mancianti F, Sozzi S. Isolation of Leishmania from a newborn puppy. Trans R Soc Trop Med Hyg. 1995;89:402. DOIPubMedGoogle Scholar
- Pangrazio KK, Costa EA, Amarilla SP, Cino AG, Silva TMA, Paixão TA, et al. Tissue distribution of Leishmania chagasi and lesions in transplacentally infected fetuses from symptomatic and asymptomatic naturally infected bitches. Vet Parasitol. 2009;165:327–31. DOIPubMedGoogle Scholar
- Masucci M, De Majo M, Contarino RB, Borruto G, Vitale F, Pennisi MG. Canine leishmaniasis in the newborn puppy. Vet Res Commun. 2003;27(Suppl 1):771–4. DOIPubMedGoogle Scholar
- Naucke TJ, Lorentz S. First report of venereal and vertical transmission of canine leishmaniosis from naturally infected dogs in Germany. Parasit Vectors. 2012;5:67. DOIPubMedGoogle Scholar
- Galindo-Sevilla N, Mancilla-Ramírez J. T-cell tolerance as a potential effect of congenital leishmaniasis on offspring immunity. Parasite Immunol. 2019;41:
e12540 . DOIPubMedGoogle Scholar - Adam GK, Omar SM, Ahmed MA, Abdallah TM, Ali AA. Cross-sectional study of the case-fatality rate among patients with visceral leishmaniasis infections during pregnancy in Sudan. Int J Gynaecol Obstet. 2018;140:119–20. DOIPubMedGoogle Scholar
- Akopyants NS, Kimblin N, Secundino N, Patrick R, Peters N, Lawyer P, et al. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science. 2009;324:265–8. DOIPubMedGoogle Scholar
- Inbar E, Shaik J, Iantorno SA, Romano A, Nzelu CO, Owens K, et al. Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in Leishmania. PLoS Genet. 2019;15:
e1008042 . DOIPubMedGoogle Scholar - Figueiró-Filho EA, El Beitune P, Queiroz GT, Somensi RS, Morais NO, Dorval MEC, et al. Visceral leishmaniasis and pregnancy: analysis of cases reported in a central-western region of Brazil. Arch Gynecol Obstet. 2008;278:13–6. DOIPubMedGoogle Scholar
- Pagliano P, Carannante N, Rossi M, Gramiccia M, Gradoni L, Faella FS, et al. Visceral leishmaniasis in pregnancy: a case series and a systematic review of the literature. J Antimicrob Chemother. 2005;55:229–33. DOIPubMedGoogle Scholar
- Anderson DC, Buckner RG, Glenn BL, MacVean DW. Endemic canine leishmaniasis. Vet Pathol. 1980;17:94–6. DOIPubMedGoogle Scholar
- Gaskin AA, Schantz P, Jackson J, Birkenheuer A, Tomlinson L, Gramiccia M, et al. Visceral leishmaniasis in a New York foxhound kennel. J Vet Intern Med. 2002;16:34–44. DOIPubMedGoogle Scholar
- Owens SD, Oakley DA, Marryott K, Hatchett W, Walton R, Nolan TJ, et al. Transmission of visceral leishmaniasis through blood transfusions from infected English foxhounds to anemic dogs. J Am Vet Med Assoc. 2001;219:1076–83. DOIPubMedGoogle Scholar
- Gibson-Corley KN, Hostetter JM, Hostetter SJ, Mullin K, Ramer-Tait AE, Boggiatto PM, et al. Disseminated Leishmania infantum infection in two sibling foxhounds due to possible vertical transmission. Can Vet J. 2008;49:1005–8.PubMedGoogle Scholar
- Duprey ZH, Steurer FJ, Rooney JA, Kirchhoff LV, Jackson JE, Rowton ED, et al. Canine visceral leishmaniasis, United States and Canada, 2000-2003. Emerg Infect Dis. 2006;12:440–6. DOIPubMedGoogle Scholar
- Schantz PM, Steurer FJ, Duprey ZH, Kurpel KP, Barr SC, Jackson JE, et al. Autochthonous visceral leishmaniasis in dogs in North America. J Am Vet Med Assoc. 2005;226:1316–22. DOIPubMedGoogle Scholar
- Schaut RG, Robles-Murguia M, Juelsgaard R, Esch KJ, Bartholomay LC, Ramalho-Ortigao M, et al. Vectorborne transmission of Leishmania infantum from hounds, United States. Emerg Infect Dis. 2015;21:2209–12. DOIPubMedGoogle Scholar
- Duthie MS, Petersen C. Could canine visceral leishmaniosis take hold in the UK? Vet Rec. 2019;184:438–40. DOIPubMedGoogle Scholar
- Rosypal AC, Zajac AM, Lindsay DS. Canine visceral leishmaniasis and its emergence in the United States. [viii.] [viii.]. Vet Clin North Am Small Anim Pract. 2003;33:921–37, viii. DOIPubMedGoogle Scholar
- Larson M, Toepp A, Scott B, Kurtz M, Fowler H, Esfandiari J, et al.; EPID:158:001. Semi-quantitative measurement of asymptomatic L. infantum infection and symptomatic visceral leishmaniasis in dogs using Dual-Path Platform® CVL. Appl Microbiol Biotechnol. 2017;101:381–90. DOIPubMedGoogle Scholar
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. DOIPubMedGoogle Scholar
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33.
- Pembleton LW, Cogan NOI, Forster JW. StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol Ecol Resour. 2013;13:946–52. DOIPubMedGoogle Scholar
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–5. DOIPubMedGoogle Scholar
- Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64. DOIPubMedGoogle Scholar
- Britton T, Anderson CL, Jacquet D, Lundqvist S, Bremer K. Estimating divergence times in large phylogenetic trees. Syst Biol. 2007;56:741–52. DOIPubMedGoogle Scholar
- Franssen SU, Durrant C, Stark O, Moser B, Downing T, Imamura H, et al. Global genome diversity of the Leishmania donovani complex. eLife. 2020;9:
e51243 . DOIPubMedGoogle Scholar - Carnielli JBT, Crouch K, Forrester S, Silva VC, Carvalho SFG, Damasceno JD, et al. A Leishmania infantum genetic marker associated with miltefosine treatment failure for visceral leishmaniasis. EBioMedicine. 2018;36:83–91. DOIPubMedGoogle Scholar
- Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–47. DOIPubMedGoogle Scholar
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al.; 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. DOIPubMedGoogle Scholar
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. DOIPubMedGoogle Scholar
- Kofler R, Pandey RV, Schlötterer C. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics. 2011;27:3435–6. DOIPubMedGoogle Scholar
- Teixeira DG, Monteiro GRG, Martins DRA, Fernandes MZ, Macedo-Silva V, Ansaldi M, et al. Comparative analyses of whole genome sequences of Leishmania infantum isolates from humans and dogs in northeastern Brazil. Int J Parasitol. 2017;47:655–65. DOIPubMedGoogle Scholar
- Kuhls K, Chicharro C, Cañavate C, Cortes S, Campino L, Haralambous C, et al. Differentiation and gene flow among European populations of Leishmania infantum MON-1. PLoS Negl Trop Dis. 2008;2:
e261 . DOIPubMedGoogle Scholar - Leblois R, Kuhls K, François O, Schönian G, Wirth T. Guns, germs and dogs: On the origin of Leishmania chagasi. Infect Genet Evol. 2011;11:1091–5. DOIPubMedGoogle Scholar
- Dumetz F, Imamura H, Sanders M, Seblova V, Myskova J, Pescher P, et al. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio. 2017;8:e00599–17. DOIPubMedGoogle Scholar
- Franssen SU, Takele Y, Adem E, Sanders MJ, Müller I, Kropf P, et al. Diversity and within-host evolution of Leishmania donovani from visceral leishmaniasis patients with and without HIV coinfection in northern Ethiopia. MBio. 2021;12:
e0097121 . DOIPubMedGoogle Scholar - Naucke TJ, Amelung S, Lorentz S. First report of transmission of canine leishmaniosis through bite wounds from a naturally infected dog in Germany. Parasit Vectors. 2016;9:256. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: May 12, 2022
1These senior authors contributed equally to this article.
Table of Contents – Volume 28, Number 6—June 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Susanne Franssen, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SA, UK
Top