Volume 11, Number 3—March 2005
Research
Rapid Identification of Emerging Pathogens: Coronavirus
Cite This Article
Citation for Media
Abstract
We describe a new approach for infectious disease surveillance that facilitates rapid identification of known and emerging pathogens. The process uses broad-range polymerase chain reaction (PCR) to amplify nucleic acid targets from large groupings of organisms, electrospray ionization mass spectrometry for accurate mass measurements of PCR products, and base composition signature analysis to identify organisms in a sample. We demonstrate this principle by using 14 isolates of 9 diverse Coronavirus spp., including the severe acute respiratory syndrome–associated coronavirus (SARS-CoV). We show that this method could identify and distinguish between SARS and other known CoV, including the human CoV 229E and OC43, individually and in a mixture of all 3 human viruses. The sensitivity of detection, measured by using titered SARS-CoV spiked into human serum, was ≈1 PFU/mL. This approach, applicable to the surveillance of bacterial, viral, fungal, or protozoal pathogens, is capable of automated analysis of >900 PCR reactions per day.
Nucleic acid tests for infectious diseases are primarily based on amplification methods that use primers and probes designed to detect specific organisms. Because prior knowledge of nucleic acid sequence information is required to develop these tests, they are not able to identify unanticipated, newly emergent, or previously unknown infectious organisms. Thus, the discovery of new infectious organisms still relies largely on culture methods and microscopy, which were as important in the recent identification of the severe acute respiratory syndrome–associated coronavirus (SARS-CoV) as they were in the discovery of HIV 2 decades ago (1–4).
Broad-range polymerase chain reaction (PCR) methods provide an alternative to single-agent tests. By amplifying gene targets conserved across groups of organisms, broad-range PCR has the potential to generate amplification products across entire genera, families, or as with bacteria, an entire domain of life. This strategy has been successfully used with consensus 16S ribosomal RNA primers for determining bacterial diversity, both in environmental samples (5) and in natural human flora (6). Broad-range priming has also been described for detection of several viral families, including CoV (7), enteroviruses (8), retroid viruses (9), and adenoviruses (10). The drawback of this approach for epidemiologic applications is that the analysis of PCR products for mixed amplified samples requires sequencing hundreds of colonies per reaction, which is impractical to perform rapidly or on large numbers of samples. New approaches to the parallel detection of multiple infectious agents include multiplexed PCR methods (11,12) and microarray strategies (13–15). Microarray strategies are promising because undiscovered organisms might be detected by hybridization to probes designed to conserved regions of known families of bacteria and viruses.
We present an alternative approach for rapid, sensitive, and high-throughput detection of infectious organisms. We use broad-range PCR to generate amplification products from the broadest possible grouping of organisms, followed by electrospray ionization mass spectrometry and base composition analysis of the products (16,17). The base compositions of strategically selected regions of the genome are used to identify and distinguish organisms in the sample. Enhanced breadth of priming is achieved through the use of primers and probes containing 5-propynyl deoxycytidine and deoxyuridine nucleotides that offer increased affinity and base pairing selectivity (18,19). Positioning the 5-propynyl primidine-modified nucleotides at highly conserved positions enables priming at short consensus regions and significantly increases the extent to which broad groups of organisms can be amplified.
CoV Isolates and Broad-range PCR Primer Pairs
Table 1 lists all the CoV used in this study. Multiple sequence alignments of all available CoV nucleotide sequences from GenBank were scanned to identify pairs of potential PCR priming loci. Two target regions were selected in CoV ORF-1b (annotations based on Snijder et al. [20]), 1 in RNA-dependent RNA polymerase (RdRp) and the other in Nsp14 (Table 2). 5′ propynyl-modified pyrimidine nucleotides (shown in bold) were positioned at universally conserved positions within these primers to extend the breadth of broad-range priming to allow efficient PCR from all CoV species tested.
For each primer region, a database of expected base compositions (A, G, C, and T base counts) from all known CoV sequences in GenBank was generated (data not shown) and used in the identification and classification of the test isolates. Several of the isolates used in this study did not have a genome sequence record in GenBank. Experimentally measured base compositions from these isolates were independently verified by sequencing ≈500 base pair (bp) regions that flanked both target regions used in this study (GenBank accession nos.AY874541 and AY878317-AY878324).
RNA Extraction, Reverse Transcription, and PCR
RNA was isolated from 250 μL of CoV-infected cells or culture supernatant spiked with 3 μg of sheared poly-A DNA using Trizol or Trizol LS, respectively (Invitrogen Inc., Carlsbad, CA, USA) according to the manufacturer’s protocol. Reverse transcription was performed by mixing 10 μL of the purified RNA with 5 μL of water treated with diethyl pyrocarbonate (DEPC, Sigma-Aldrich Co., St. Louis, MO, USA) containing 500 ng random primers, 1 μg of sheared poly-A DNA, and 10 U SUPERaseIn (Ambion Inc., Woodlands, TX, USA). The mixture was heated to 60°C for 5 min and then cooled to 4°C. Following the annealing of the random primers to the RNA, 20 μL of first-strand reaction mix consisting of 2x first-strand buffer (Invitrogen Inc.), 10 mmol/L DTT, 500 µmol/L deoxynucleoside triphosphates (dNTPs), and 7.5 U SuperScript II was added to the RNA primer mixture. The RNA was reversed transcribed for 45 min at 45°C. Various dilutions of the reverse transcription reaction mixes were used directly in the PCR reactions.
All PCR reactions were performed in 50 μL with 96-well microtiter plates and M.J. Dyad thermocyclers (MJ Research, Waltham, MA, USA). The PCR reaction buffer consisted of 4 U Amplitaq Gold (Applied Biosystems, Foster City, CA USA), 1x buffer II (Applied Biosystems, Foster City, CA, USA), 2.0 mmol/L MgCl2, 0.4 mol/L betaine, 800 μmol/L dNTP mix, and 250 nmol/L propyne-containing PCR primers. The following PCR conditions were used to amplify CoV sequences: 95°C for 10 min followed by 50 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 30 s. After PCR, the amplified products were desalted before analysis by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICR-MS) by methods described previously (21). A small oligonucleotide SH2 (CGTGCATGGCGG, Synthetic Genetics, San Diego, CA, USA) was added as an internal mass standard (22); the final concentration of SH2 was 50 nmol/L.
Mass Spectrometry and Signal Processing
The mass spectrometer used in this work is based on a Bruker Daltonics (Billerica, MA, USA) Apex II 70e ESI-FTICR-MS that used an actively shielded 7 Tesla superconducting magnet. All aspects of pulse sequence control and data acquisition were performed on a 1.1 GHz Pentium II data station running Bruker’s Xmass software (Bruker Daltonics). Inputs to the signal processor are the raw mass spectra for each of the parallel PCR reactions used to analyze a single sample. The ICR-2LS software package (23) was used to deconvolute the mass spectra and calculate the mass of the monoisotopic species using an “averagine” fitting routine (24) modified for DNA (Drader et al., unpub. data). Using this approach, monoisotopic molecular weights were calculated. The spectral signals were algorithmically processed to yield base composition data as described previously (25). The amplitudes of the spectra are calibrated to indicate the number of molecules detected in the mass spectrometer; m/z and the m/z values are corrected by using internal mass standards. The algorithm computes the organism’s identity and abundances consistent with observations over all the PCR reactions run on the input sample.
Detection of Individual CoV Isolates
Figure 1

Figure 1. . Electrospray ionization Fourier transfer ion cyclotron resonance (ESI-FTICR) mass spectrum from the polymerase chain reaction (PCR) amplicons from the severe acute respiratory syndrome (SARS)-associated coronavirus obtained with the propynylated RNA-dependent...
For broad-range detection of all CoV, the 2 PCR primer target regions shown in Table 2 were used against each virus listed in Table 1. Resultant products were desalted and analyzed by FTICR-MS by methods described previously (21). The spectral signals were algorithmically processed to yield base composition data. Figure 1 shows a schematic representation of electrospray ionization, strand separation, and the actual charge state distributions of the separated sense and antisense strands of the PCR products from the RdRp primer pair for SARS-CoV. Due to the accuracy of FTICR-MS (mass measurement error ± 1 ppm), all detected masses could be unambiguously converted to the base compositions of sense and antisense strands (25).
One of the limitations of all molecular methods for detecting pathogens, including the one described here, is that unexpected variations in PCR primer target sequences in unknown species can lead to missed detection. To minimize this possibility, the primers designed in this study were selected on the basis of highly conserved regions identified by multiple sequence alignments of all previously known CoV species sequences. Further, we chose 2 amplification targets for redundant detection of the CoV and to have increased resolution to distinguish the different viral species. Both primer pairs were tested against multiple isolates from the 3 previously known CoV species groups and from SARS-CoV isolates.
The results from analysis of 14 CoV isolates are shown in Table 1. For both target regions, the measured signals agreed with compositions expected from the known CoV sequences in GenBank (data not shown). Several of the isolates used in this study did not have a genome sequence record in GenBank. Nevertheless, we were able to amplify all test viruses and experimentally determine their base compositions. These experimentally determined base compositions were confirmed by sequencing (data not shown). Thus the strategy described here enables identification of organisms without the need for prior knowledge of the sequence, provided that the broad range primers do not fail to amplify the target because of excessive numbers of mismatches.
Multiple CoV Isolates in Mixture
Figure 2
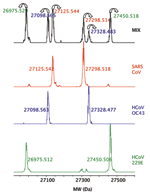
Figure 2. . Detection of 3 human coronavirus (CoV) in a mixture. The deconvoluted (neutral mass) mass spectra obtained for the RNA-dependent RNA polymerase primer for the 3 human CoV, HCoV-229E, HCoV-OC43, and...
To demonstrate the potential to detect multiple viruses in the same sample, as might occur during a coinfection, we pooled the viral extracts from 3 human CoV, HCoV-229E, HCoV-OC43, and SARS-CoV, and analyzed the mixture. Signals from all 3 viruses were clearly detected and resolved in the mass spectra (Figure 2), which demonstrated that coinfections of >1 CoV species could be identified. We have previously determined that the system can reliably detect multiple species in ratios of ≈1:1000, while varying input loads from 10 to 10,000 organisms (data not shown).
Sensitivity
To determine sensitivity in a clinical sample, viable, titered SARS-CoV was added to human serum and analyzed in 2 ways. In the first, RNA was isolated from serum containing 2 concentrations of the virus (1.7 x 105 and 170 PFU/mL), reverse transcribed to cDNA with random primers, and serially diluted (10-fold), before PCR amplification with both RdRp and Nsp14 primer sets. By using this approach, the assay was sensitive to ≈102 PFU per PCR reaction (≈1.7 PFU/mL serum). We estimated the number of reverse-transcribed SARS genomes by competitive, quantitative PCR with a nucleic acid internal standard (data not shown). Analysis of ratios of mass spectral peak heights of titrations of the internal standard and the SARS cDNA showed that ≈300 reverse-transcribed viral genomes were present per PFU, similar to the ratio of viral genome copies per PFU previously reported for RNA viruses (26). By using this estimate, PCR primers were sensitive to 3 genome equivalents per PCR reaction, which is consistent with previously reported detection limits for optimized SARS-specific primers (27,28). In the second method, we spiked 10-fold dilutions of the SARS virus into serum before RT-PCR and could reliably detect 1 PFU (≈300 genomes) per PCR reaction or 170 PFU (5.1 x 104 genomes) per mL serum. The discrepancy between the detection sensitivities in the 2 experimental protocols described above suggests that losses were associated with RNA extraction and reverse transcription when very little virus was present (<300 genome copies) in the starting sample in serum. This finding is consistent with results for direct measurement of RNA viruses from patient samples (26). Therefore, in a practical experimental analysis of a tissue sample, the limit of sensitivity was ≈1 PFU per PCR reaction.
RNA Virus Classification with Base Compositions
We have described a novel approach using base composition analysis for viral identification. However, since RNA virus nucleotide sequences mutate over time within the functional constraints allowed by selection pressure (29), the utility of this method to correctly classify RNA viruses depends on the resolution needed for a particular application. We considered 2 specific applications. The first was to distinguish SARS-CoV from other species of CoV that infect humans, namely HCoV-OC43 and HCoV-229E. The second application was the utility of the technique for exploration of animal reservoirs for the discovery of SARS-related CoV species.
Figure 3
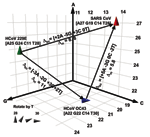
Figure 3. . Spatial representation of base compositions for the 3 coronavirus (CoV) species known to infect humans. Severe acute respiratory syndrome (SARS), HCoV-OC43, and HCoV-229E base compositions in the region amplified by...
To quantitatively analyze the resolving power of base compositions, we mathematically modeled base composition variations using known sequences of multiple isolates of hepatitis C virus (HCV) in GenBank (H. Levene, et al., unpub. data). HCV sequence-derived mutation probabilities were used to estimate the extent of base composition variations for CoV species. Figure 3 shows a plot of the base compositions for the RdRp target region for the 3 CoV known to infect humans. Δbc represents the net changes in composition required for strain variants of 229E or OC43 to be misidentified as SARS, and Δm the probability of occurrence of these changes. The cumulative probability of misclassifying either 229E or OC43 as SARS by using base composition measurements from both target regions was low (Δm >10), even allowing for unseen variations in those 2 viruses. Thus, for use in human clinical diagnostics, base composition analysis of the 2 target regions described here would provide corroborative information and accurate species identification of CoV infections.
Figure 4
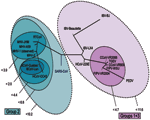
Figure 4. . Representation of the mutational distances calculated for the 2 selected primer sets overlaid on the coronavirus phylogenetic tree. Each oval represents grouping of members contained within it; numbers next to...
To determine the utility of base composition analysis in the search for animal CoV species, we calculated the cumulative mutation distances for both target regions for all known CoV and plotted groups where all members fall within certain probability thresholds, as shown in Figure 4. A series of nested ovals represents subgroupings of species, where the maximal distance between known members of a subgroup is represented by the Δm next to the oval. By using the above classification metric, SARS-CoV would be considered the first member of a new group of CoV, not a member of the core group 2 cluster, although it would be placed closest to group 2 (Δm < 10.2). These findings are similar to those recently described by Snijder et al., who used sequence data from the replicase genes (5,487 bp) in ORF1b and suggested that the SARS-CoV was most closely related to and possibly an early split-off from group 2 CoV (20). However, substantial space exists around SARS-CoV where as yet undiscovered SARS-CoV could populate a subgroup without being confused with the group 2 or other CoV.
The strategy we describe allows rapid identification of new viral species members of previously characterized viral families, without the need for prior knowledge of their sequence, through use of integrated electrospray ionization mass spectrometry and base composition analysis of broad-range PCR products. Broad-range PCR reactions are capable of producing products from groups of organisms, rather than single species, and the information content of each PCR reaction is potentially very high. Further, in many cases, including the SARS-CoV detection described in this article, priming across broadly conserved regions provides adequate species detection and taxonomic resolution. In cases where additional subspecies level classification becomes important, broad primers can be followed up with more species-specific primers that can detect even single nucleotide changes (SNPs) or alternatively, larger regions of the identified species can be analyzed by sequencing. Despite the advances in high throughput sequencing, however, it is impractical as a front-end detector in a routine survey and detection setting. The mass spectrometer is capable of analyzing complex PCR products at a rate of ≈1 minute per sample. Because the process is performed in an automated, microtiter plate format, large numbers of samples can be examined (>900 PCR reactions/day/instrument), which makes this process practical for large-scale analysis of clinical or environmental surveillance samples in public health laboratory settings. The current generations of ESI mass spectrometers used in the detector cost approximately U.S.$150,000 and can be operated 24 hours per day by trained technicians. Tools for analyzing mass spectrometry data are widely available and are described in detail elsewhere (23–25). A comparable alternative to the methods described here are microarrays, which can also provide broad range detection.
This approach can be extended to other viral, bacterial, fungal, or protozoal pathogen groups and is a powerful new paradigm for timely identification of previously unknown organisms that cause disease in humans or animals and for monitoring the progress of epidemics.
Dr. Sampath is the director of Genomics and Computational Biology at Ibis Therapeutics, a division of Isis Pharmaceuticals, Inc. He leads Ibis’ genomics efforts in microbial detection and diagnosis. His research interests include pathogen discovery, epidemiologic surveillance, and clinical infectious diagnostics.
Acknowledgment
This study used platform technology that was funded in part by the Department of Defense through its DARPA Special Projects Office for the development of TIGER Technology, under contract MDA972-00-C-0053; patents are currently pending in the United States and internationally. Additional funding and support was provided by grants from the National Institutes of Health, AI 25913 to M.B., and T32 NS 41219 to B.N. All authors, with the exception of M.B. and B.N., are employees and stockholders of Isis Pharmaceuticals, Inc.
References
- Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K, Identification of severe acute respiratory syndrome in Canada. N Engl J Med. 2003;348:1995–2005. DOIPubMedGoogle Scholar
- Peiris JS, Lai S, Poon L, Guan Y, Yam L, Lim W, Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–25. DOIPubMedGoogle Scholar
- Falsey AR, Walsh EE. Novel coronavirus and severe acute respiratory syndrome. Lancet. 2003;361:1312–3. DOIPubMedGoogle Scholar
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–66. DOIPubMedGoogle Scholar
- Schmidt TM, DeLong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 1991;173:4371–8.PubMedGoogle Scholar
- Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci U S A. 1999;96:14547–52. DOIPubMedGoogle Scholar
- Stephensen CB, Casebolt DB, Gangopadhyay NN. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 1999;60:181–9. DOIPubMedGoogle Scholar
- Oberste MS, Maher K, Pallansch MA. Molecular phylogeny and proposed classification of the simian picornaviruses. J Virol. 2002;76:1244–51. DOIPubMedGoogle Scholar
- Mack DH, Sninsky JJ. A sensitive method for the identification of uncharacterized viruses related to known virus groups: hepadnavirus model system. Proc Natl Acad Sci U S A. 1988;85:6977–81. DOIPubMedGoogle Scholar
- Echavarria M, Forman M, Ticehurst J, Dumler A, Charache P. PCR method for detection of adenovirus in urine of healthy and human immunodeficiency virus-infected individuals. J Clin Microbiol. 1998;36:3323–6.PubMedGoogle Scholar
- Fout GS, Martinson BC, Moyer MW, Dahling DR. A multiplex reverse transcription-PCR method for detection of human enteric viruses in groundwater. Appl Environ Microbiol. 2003;69:3158–64. DOIPubMedGoogle Scholar
- Brito DA, Ramirez M, de Lencastre H. Serotyping Streptococcus pneumoniae by multiplex PCR. J Clin Microbiol. 2003;41:2378–84. DOIPubMedGoogle Scholar
- Wilson KH, Wilson WJ, Radosevich JL, DeSantis TZ, Viswanathan VS, Kuczmarski TA, High-density microarray of small-subunit ribosomal DNA probes. Appl Environ Microbiol. 2002;68:2535–41. DOIPubMedGoogle Scholar
- Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, Microarray-based detection and genotyping of viral pathogens. Proc Natl Acad Sci U S A. 2002;99:15687–92. DOIPubMedGoogle Scholar
- Wang D, Urisman A, Liu YT, Springer M, Ksiazek TG, Erdman D, Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003;1:257–60. DOIPubMedGoogle Scholar
- Sampath R, Ecker DJ. Novel biosensor for infectious disease diagnostics. In: Knobler SE, Mahmoud A, Lemon S, Mack A, Sivitz L, Oberholtzer K, editors. Learning from SARS: preparing for the next disease outbreak. Washington: The National Academies Press; 2004. p. 181–5.
- Hofstadler SA, Sampath R, Blyn LB, Eshoo MW, Hall TA, Jiang Y, TIGER: the universal biosensor. Int J Mass Spectrom. 2004. [cited 22 Dec 2004]. Available from http://www.sciencedirect.com/science/article/B6VND-4F31R2G-1/2/96bc83d6dec9dfdd8429ca692be52a08
- Wagner RW, Matteucci MD, Lewis JG, Gutierrez AJ, Moulds C, Froehler BC. Antisense gene inhibition by oligonucleotides containing C-5 propyne pyrimidines. Science. 1993;260:1510–3. DOIPubMedGoogle Scholar
- Barnes TW, Turner DH. Long-range cooperativity due to C5-propynylation of oligopyrimidines enhances specific recognition by uridine of ribo-adenosine over ribo-guanosine. J Am Chem Soc. 2001;123:9186–7. DOIPubMedGoogle Scholar
- Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LLM, Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. DOIPubMedGoogle Scholar
- Jiang Y, Hofstadler SA. A highly efficient and automated method of purifying and desalting PCR products for analysis by electrospray ionization mass spectrometry. Anal Biochem. 2003;316:50–7. DOIPubMedGoogle Scholar
- Greig M, Griffey RH. Utility of organic bases for improved electrospray mass spectrometry of oligonucleotides. Rapid Commun Mass Spectrom. 1995;9:97–102. DOIPubMedGoogle Scholar
- Anderson GA, Bruce JE. ICR2LS. Richland (WA): Pacific Northwest National Laboratory; 1995.
- Senko MW, Beu SC, McLafferty FW. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J Am Soc Mass Spectrom. 1995;6:229–33. DOIGoogle Scholar
- Muddiman DC, Anderson GA, Hofstadler SA, Smith RD. Length and base composition of PCR-amplified nucleic acids using mass measurements from electrospray ionization mass spectrometry. Anal Chem. 1997;69:1543–9. DOIPubMedGoogle Scholar
- Towner JS, Rollin PE, Bausch DG, Sanchez A, Crary SM, Vincent M, Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J Virol. 2004;78:4330–41. DOIPubMedGoogle Scholar
- Drosten C, Gunther S, Preiser W, Van Der Werf S, Brodt HR, Becker S, Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. DOIPubMedGoogle Scholar
- Nitsche A, Schweiger B, Ellerbrok H, Niedrig M, Pauli G. SARS coronavirus detection. Emerg Infect Dis. 2004;10:1300–4.PubMedGoogle Scholar
- Jenkins GM, Rambaut A, Pybus OG, Holmes EC. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol. 2002;54:156–65. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 11, Number 3—March 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Rangarajan Sampath, Ibis Therapeutics, 1891 Rutherford Ct., Carlsbad, CA 92008, USA; fax: 760-603-4653
Top