Volume 29, Number 2—February 2023
Research
Novel Prion Strain as Cause of Chronic Wasting Disease in a Moose, Finland
Cite This Article
Citation for Media
Abstract
Our previous studies using gene-targeted mouse models of chronic wasting disease (CWD) demonstrated that Norway and North America cervids are infected with distinct prion strains that respond differently to naturally occurring amino acid variation at residue 226 of the prion protein. Here we performed transmissions in gene-targeted mice to investigate the properties of prions causing newly emergent CWD in moose in Finland. Although CWD prions from Finland and Norway moose had comparable responses to primary structural differences at residue 226, other distinctive criteria, including transmission kinetics, patterns of neuronal degeneration, and conformational features of prions generated in the brains of diseased mice, demonstrated that the strain properties of Finland moose CWD prions are different from those previously characterized in Norway CWD. Our findings add to a growing body of evidence for a diverse portfolio of emergent strains in Nordic countries that are etiologically distinct from the comparatively consistent strain profile of North America CWD.
Prions are infectious proteins that cause fatal, incurable neurodegenerative diseases of humans and animals, which include Creutzfeldt-Jakob disease (CJD), sheep scrapie, bovine spongiform encephalopathy, and chronic wasting disease (CWD) of cervids. The extraordinary biology and transmissibility of these disorders stems from the protean conformational properties of the prion protein (PrP). Although the secondary structure of host-encoded cellular PrP (PrPC) is predominantly α-helical, during disease its relatively underglycosylated infectious counterpart (PrPSc) assembles into amyloid fibrils with parallel, in-register, intermolecular β-sheets (1–7). The replicative properties of prions stem from the capacity of PrPSc to template its conformation on PrPC in a cyclical process resulting in exponential accumulation of prion infectivity (8–11).
Although they lack nucleic acids, prions exhibit heritable strain properties that influence disease outcomes, including the time between infection and disease onset (incubation period), clinical signs, patterns of neuronal degeneration and PrPSc deposition in the central nervous system (CNS), and the ability to replicate in non-CNS tissues such as the lymphoreticular system and musculature (12). Strain properties also influence the capacity of prions from one species to cause disease in a different species (13). Heritable strain information appears to be enciphered by distinct PrPSc conformations that are faithfully propagated during prion replication (14,15).
The food-chain transmission of bovine spongiform encephalopathy prions that resulted in a variant form of CJD illustrates the unpredictable potential of emergent strains for zoonotic transmission (13). Although novel prion diseases and strain variants continue to arise in increasing numbers of animal species, CWD elicits particular concern (16). After its initial description in a captive deer facility (17), uncontrolled contagious transmission has resulted in growing numbers of CWD-affected cervids in at least 30 US states and 3 Canada provinces (18). Inadvertent importation of subclinically diseased animals from North America led to the establishment of CWD in South Korea (19–21). CWD was also diagnosed in free-ranging Norwegian reindeer in 2016 (22), and additional cases were subsequently identified in growing numbers of moose, red deer, and reindeer from Norway, Sweden, and Finland (23).
The development and application of genetically modified, CWD-susceptible mice expressing cervid PrPC (CerPrPC) has provided insights into multiple aspects of pathogenesis, including the impact of naturally occurring PRNP coding sequence variations (16). Whereas North America deer and moose encode glutamine (Q) at codon 226 (CerPrP-Q226), North America elk encode glutamate (E) at this position (CerPrP-E226). Early studies in transgenic (Tg) mice suggested a role for this variation in the selection and propagation of CWD strains (24–26). To precisely assess the effects of this primary structural difference, we created CWD-susceptible gene-targeted (Gt) mice in which the PrP coding sequence was replaced with CerPrP-Q226 or CerPrP-E226 (27). Because the resulting mice, referred to as GtQ and GtE mice, express equivalent, physiologically controlled levels of CerPrPC and are otherwise syngeneic, we were able to ascribe distinct disease outcomes in each line to the effects of these amino acids (27). Furthermore, in contrast to previous CWD-susceptible Tg mice, Gt mice recapitulated the lymphotropic properties of CWD strains (27,28). By using Gt mice, we showed that emergent strains causing CWD in Norway reindeer and moose were unrelated to established North America forms of CWD and that they responded differently to variation at residue 226 (28). These findings underscored the utility of Gt mice for accurately defining the strain properties of emergent CWD prions (28). The goal of this study was to characterize a newly emergent form of CWD in Finland moose.
Ethics Statement
We performed all animal work in an Association for Assessment for Accreditation of Laboratory Animal Care–accredited facility in accordance with the Guide for the Care and Use of Laboratory Animals (https://www.ncbi.nlm.nih.gov/books/NBK54050). All procedures used in this study were performed in compliance with and were approved by the Colorado State University Institutional Animal Care and Use Committee.
Isolates
The Finland moose (M-F1) CWD isolate was from a free-ranging female moose identified to be sick and later found dead in 2018. The North America moose (M-US1) CWD isolate was from a captive moose orally inoculated with a pooled preparation of deer CWD. North America elk CWD isolate (E-US1), and Norway moose CWD isolates (M-NO1, M-NO2, and M-NO3) have been described previously (28).
Mouse Models and Incubation Time Assay
Development and characterization of Tg mice expressing cervid PrP with glutamine at residue 226 (TgQ), Tg mice expressing cervid PrP with glutamate at residue 226 (TgE), GtQ and GtE mice (expressing the same thing as Tg mice), and the prion incubation time assay have been described previously (24,27–29). We used equal numbers of male and female mice in inoculation studies. We confirmed clinical diagnoses of prion disease by PrPSc detection in the CNS and neuropathologic assessments.
Analysis of PrPSc by Western Blotting
We performed analysis of PrPSc by western blotting as described previously (27,28). Equivalent amounts of protein were treated with 50 μg/mL of proteinase K (PK; Roche, https://www.roche.com) in the presence of 2% sarkosyl for 1 hour at 37°C. We performed sodium dodecyl sulfate–polyacrylamide gel electrophoresis by using precast 12% discontinuous Bis-Tris gels (Bio-Rad Laboratories, Inc., https://www.bio-rad.com). We detected proteins transferred to Immobilon-FL PVDF membranes (Millipore, https://www.emdmillipore.com) with monoclonal antibody (mAb) PRC5 at a dilution of 1:5,000 and mAb PRC1 at a dilution of 1:2,500 (30), followed by horseradish peroxidase–conjugated anti-mouse IgG secondary antibody. We developed membranes by using ECL 2 Western Blotting Substrate (ThermoFisher Scientific, https://www.thermofisher.com).
Conformational Stability Assay
We conducted a conformational stability assay as described previously (27,28). We detected PrPSc on membranes with mAb PRC5 (30) at a dilution of 1:5,000, followed by horseradish peroxidase–conjugated goat anti-mouse IgG secondary antibody. We developed membranes with ECL 2 Western Blotting Substrate and scanned them with an ImageQuant LAS 4000 digital camera system (GE Healthcare, https://www.gehealthcare.com) and analyzed signals with ImageQuant TL 7.0 software (GE Healthcare).
Histoblot Analysis
We performed histoblot analysis as described previously (31). We detected PrPSc on membranes by using mAb PRC5 at a dilution of 1:5,000, followed by alkaline phosphatase conjugated goat anti-mouse IgG (Southern Biotech, https://www.southernbiotech.com) at a dilution of 1:5,000. We developed membranes by using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (Sigma Aldrich, https://www.sigmaaldrich.com) and captured images with a Nikon SMZ1000 microscope (https://www.microscope.healthcare.nikon.com).
Neuropathologic and Immunohistochemical Analyses
We dissected brains rapidly after euthanizing the animal and immersion fixed tissues in 10% buffered formalin. We embedded tissues in paraffin and mounted 8 μm–thick coronal microtome sections onto positively charged glass slides. We performed immunohistochemical analyses of PrP in brain sections by using mAb D18 as described previously (28).
Neuropathologic Lesion Profiling
We sectioned paraffin-embedded mouse brains coronally to areas corresponding to 5 levels of the brain containing the 9 mouse brain regions of interest. We captured images of hematoxylin and eosin–stained sections by using a Photometrics CoolSNAP digital camera (https://www.photometrics.com) and an Olympus BX51 fluorescence microscope (https://www.olympus-lifescience.com) and analyzed them by using Olympus cellSens imaging software (standard version). We assessed the severity of vacuolar degeneration in cerebral gray matter from 9 regions, as described previously (32), by using a team of 4 investigators, who scored the images on scale of 0 to 4, with 4 corresponding to severe vacuolation.
Statistical Analyses
We performed statistical analyses by using GraphPad Prism 8.0 software (https://www.graphpad.com). We assessed statistical significance between survival curves of inoculated groups by comparing median times of survival of various inoculated groups using the log-rank (Mantel-Cox) test. We assessed statistical significance between denaturation curves of PrPSc by comparing log half maximal effective concentration values.
Figure 1

Figure 1. Transmission properties of Finland moose chronic wasting disease (CWD) prions in GtQ and GtE mice. Survival curves of intracerebrally inoculated GtQ and GtE mice are shown. A–C) Primary transmissions; D–F)...
We registered disease after ≈260 days in all but 1 of 11 GtQ mice inoculated with a homogenate of CNS from the index case of Finland CWD, M-F1 (Table 1; Figure 1, panel A). Brain homogenates from 2 diseased GtQ mice, referred to as GtQ (M-F1) brain 1 and brain 2 (Figure 1, panel A), produced disease after ≈225 days following intracerebral inoculation of additional GtQ mice (Table 2; Figure 1, panel D, E). By contrast, all GtE mice inoculated with M-F1 brain homogenate remained free of disease for up to 545 days (Table 1; Figure 1, panel A), and GtQ (M-F1) brain 1 produced disease in only 4 of 7 inoculated GtE mice after ≈470 days (Table 2; Figure 1, panel D), whereas GtQ (MF-1) brain 2 failed to produced disease in GtE mice after >510 days (Table 2; Figure 1, panel E). We conclude that M-F1 prion replication is supported when Q is encoded at residue 226 of host-encoded PrP (CerPrPC-Q226) and relatively restricted when E is present at that position. In support of this conclusion, whereas CNS overexpression of CerPrPC-Q226 produced faster times to disease in TgQ compared with GtQ mice, only a single inoculated TgE mouse had disease after ≈390 days (Appendix Table 1, Figure 1, panel A).
Figure 2

Figure 2. Western blot analyses of cervid prion protein (PrP) scrapie in mice infected with Finland, Norway, and North America chronic wasting disease (CWD) isolates. A, B) Western blots of CNS homogenates...
Figure 3
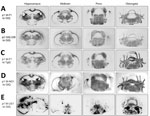
Figure 3. Global central nervous system distribution of cervid prion protein scrapie in Tg and Gt mice infected with Finland, Norway, and North America chronic wasting disease (CWD) cryostat coronal brain sections...
Figure 4

Figure 4. Lesion profiling in GtQ mice infected with Finland and Norway moose chronic wasting disease (CWD) isolates. Lesion profiles in groups of GtQ mice (CWD-susceptible gene-targeted mice in which the prion...
Figure 5
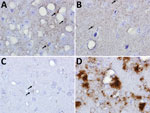
Figure 5. Immunohistochemical analyses of disease-associated prion protein (PrP) in the CNS of transgenic (Tg) and gene-targeted (Gt) mice infected with Finland, Norwway, and North America chronic wasting disease (CWD) isolates. A)...
We confirmed clinical diagnoses of prion disease by using western blotting of PK-resistant CerPrPSc in brain homogenates (Figure 2, panels A, C), histoblotting of CerPrPSc in coronal sections (Figure 3, panels A–C), and microscopic analyses of neuronal vacuolation (Figure 4; Appendix Figure 3, panel B) and CerPrPSc deposition in the CNS (Figure 5, panels A, B). Although spleen homogenates of GtQ mice infected with North America CWD and Norway reindeer CWD prions contained CerPrPSc, there was no evidence of prion replication in the spleens of GtQ mice after primary or secondary passage of the M-F1 isolate, or, as observed previously (28), in the spleens of GtQ mice infected with Norway moose CWD isolate M-NO1 (Figure 2, panel E). We conclude that whereas North America and Norway reindeer CWD prions are lymphotropic, Finland and Norway moose CWD prions are nonlymphotropic. In support of this conclusion, neither Gt nor Tg mice had disease after intracerebral inoculation with lymphoid tissue homogenates from M-F1 (Figure 1, panel B; Appendix Figure 1, panel B).
We questioned whether the inefficient transmission of M-F1 prions in GtE and TgE mice could be overcome by adaptation in a CerPrP-E226 background. Serial passage of M-F1 prions from the diseased TgE mouse, referred to as TgE (M-F1) (Figure 2, panel A; Appendix Table 1, Figure 1, panel A), produced disease in GtQ mice after ≈230 days but failed to produce disease in GtE mice after ≈500 days (Appendix Table 2, Figure 1, panel E). Because serial passage of M-F1 prions from diseased TgQ mice also produced disease in GtQ mice after ≈230 days and failed to produce disease in GtE mice after ≈500 days (Appendix Table 2, Figure 1, panel D), we conclude that the M-F1 strain favors conformational conversion of CerPrPC-Q226 over CerPrPC-E226 regardless of whether prions are composed of CerPrPSc-E226 or CerPrPSc-Q226.
Our previous studies showed that propagation of prions from 3 Norway moose CWD isolates, referred to as M-NO1, M-NO2, and M-NO3, was also supported by expression of CerPrPC-Q226 and relatively restricted by CerPrPC-E226 (28). Despite this shared feature, comparisons of primary transmissions to GtQ mice showed that the mean incubation time of M-F1 was faster than those of M-NO1, M-NO2, or M-NO3 (p<0.0001) (Table 1; Figure 1, panel C). Primary transmission of M-F1 was also faster than Norway moose CWD isolates in TgQ mice (p>0.0001) (Appendix Table 1, Figure 1, panel C). We also observed faster disease onsets when serial transmissions of GtQ (M-F1) brains 1 and 2 were compared with serially transmitted M-NO1 and M-NO2 in GtQ mice (Table 2; Figure 1, panel F) and when M-F1 and Norway moose CWD prions were serially propagated from TgQ to GtQ mice (Appendix Table 2, Figure 1, panel F). Those consistently different disease kinetics during primary and secondary transmissions are indicative of strain differences between Finland and Norway moose CWD prions.
To substantiate that conclusion, we assessed additional strain features in infected GtQ mice. Semiquantitative assessment by brain lesion profiling (32) showed that the extent and regional distribution of spongiform degeneration varied depending on whether GtQ mice were infected with Finland or Norway moose CWD prions (Figure 4). Differences between GtQ mice infected with M-F1 and M-NO1 were particularly evident in the septum, retrosplenial and adjacent motor cortex, and cingulate and adjacent motor cortex. In these areas the intensity of spongiform degeneration was more pronounced in GtQ mice infected with M-F1 (Figure 4; Appendix Figure 3, panel A, B). The distinctive lesion profile of M-F1 was sustained after serial passage to additional GtQ mice (Figure 4).
Figure 6
![Responses of Finland, Norway, and North America moose chronic wasting disease (CWD) prions propagated in CWD-susceptible mice to increasing concentrations of GdnHCl. A) Responses of CerPrPSc in central nervous system (CNS) of diseased GtQ mice to proteinase K treatment after denaturation with increasing concentrations of GdnHCl. Magenta symbols indicate M-F1 CWD, green symbols M-NO1 CWD, and blue symbols M-US1 CWD. B) Denaturation profiles of cervid PrP scrapie in CNS of GtQ mice infected with M-F1. Magenta squares and solid lines depict passage 1; dotted magenta squares and dashed lines depict GtQ (M-F1) brains 1 and 2 (passage 2). C) Denaturation profiles of cervid PrP scrapie in the CNS of TgQ mice infected with M-F1 (magenta circles), TgQ mice infected with M-F1 passaged through TgE mice [TgE (M-F1)] (orange circles), and TgQ mice infected with M-F1 passaged through TgQ mice [TgQ (M-F1)] (dotted magenta circles). Proteinase K–resistant PrPSc was quantified by densitometry of immunoprobed dot blots and plotted against GdnHCl concentration. Sigmoidal dose-response curves were plotted using a 4-parameter algorithm and nonlinear least-square fit. Error bars indicate +SEM of data from analyses of >3 animals per group. GdnHCl1/2 values (mol/L) for each infection are reported on the right-hand side of each graph. Significance calculated by pairwise analysis of GdnHCl1/2 values from best fit curves. Fapp, fraction of apparent PrPSc = (maximum signal-individual signal)/(maximum signal-minimum signal); GdnHCl, guanidine hydrochloride; GdnHCl1/2, guanidine hydrochloride half-maximal denaturation; GtQ, CWD-susceptible gene-targeted mice in which the prion protein coding sequence was replaced with one encoding glutamine at codon 226; NS, not significant; PK, proteinase K; PrP, prion protein; TgE, transgenic mice expressing cervid PrP with glutamate at residue 226; TgQ, transgenic mice expressing cervid PrP with glutamine at residue 226.](/eid/images/22-0882-F6-tn.jpg)
Figure 6. Responses of Finland, Norway, and North America moose chronic wasting disease (CWD) prions propagated in CWD-susceptible mice to increasing concentrations of GdnHCl. A) Responses of CerPrPScin central nervous...
We assessed the responses of CerPrPSc produced in the brains of mice infected with Finland, Norway, and North America moose CWD prions to denaturation with increasing concentrations of guanidine hydrochloride (GdnHCl) (Figure 6). This measure of PrPSc stability is associated with conformational variation among prion strains (33). Denaturation profiles and concentrations of GdnHCl producing half-maximal denaturation indicated that the stability of CerPrPSc in GtQ mice infected with M-F1 was equivalent to CerPrPSc in the brains of GtQ mice infected with North America moose CWD and lower than that of CerPrPSc in the brains of GtQ mice infected with M-NO1 (p<0.0001) (Figure 6, panel A). The conformational stability of CerPrPSc produced on primary passage of M-F1 was maintained upon serial transmissions of GtQ (M-F1) brains 1 and 2 to GtQ mice (Figure 6, panel B). We also observed overlapping denaturation curves and comparable half-maximal denaturation values in the range of 2.59 to 2.95 upon infection of TgQ mice with M-F1 prions and M-F1 prions passaged in TgE or TgQ mice (Figure 6, panel C). We conclude that conformational properties of M-F1 prions are distinct from those of M-NO1 prions, they are stable during iterative passages, and they remain unchanged after passage in mice expressing CerPrPC-E226.
Despite their different transmission properties, neuropathologic profiles, and conformations, Finland and Norway CWD prions shared certain features. Western blotting showed that levels of CerPrPSc in the brains of GtQ mice infected with M-F1 and M-NO1 were lower than the levels of CerPrPSc in the brains of GtQ mice infected with North America CWD (Figure 2). This finding is consistent with previous studies showing consistently reduced CerPrPSc accumulation after Norway moose CWD prion infection (28). The electrophoretic migration profiles of CerPrPSc resulting from infection of GtQ mice with M-F1 and M-NO1 were both more rapid than CerPrPSc produced by infection with North America CWD (Figure 2, panel A). This finding also is consistent with our previous descriptions of Norway moose CerPrPSc (28). Finally, also in accordance with previous findings (28), CerPrPSc in the brains of M-F1 and M-NO1 infected mice was relatively refractory to detection by mAb PRC1 compared with CerPrPSc generated by infection with North America CWD (Figure 2, panel B). The faster migration of CerPrPSc generated by infection with M-F1 and M-NO1 and its comparative resistance to detection by PRC1 results from PK cleavage downstream from the PRC1 epitope at residue 90 in both strains (Appendix Figure 2). Serial transmissions of M-F1, M-NO1, and North America CWD prions produced CerPrPSc immunoblotting profiles consistent with primary passages (Figure 2, panel C, D). We speculate that the low levels of PRC1-reactive CerPrPSc in brain extracts of mice infected with M-F1 and M-NO1 CWD prions (Figure 2, panel B, D) correspond to minor strain components that remain obscured when probing with nondiscriminatory mAbs. Using discriminatory mAbs revealed similar coexistence of multiple PrPSc types in persons with CJD (34).
Analyses of PK-resistant CerPrPSc in histoblotted coronal brain sections revealed a diffuse, symmetric deposition pattern in GtQ mice infected with M-F1 (Figure 3, panel A) that was indistinguishable from the pattern in GtQ mice infected with M-NO1 CWD (Figure 3, panel D), and consistent with previous analyses of mice infected with Norway moose CWD (28). This pattern was recapitulated after iterative passage of GtQ (M-F1) prions to GtQ mice (Figure 3, panel B) and after infection of TgQ mice with M-F1 (Figure 3, panel C). This diffuse pattern differed from the disorganized, asymmetrically distributed amalgamations of CerPrPSc produced by infection of GtQ mice with North America CWD prions (Figure 3, panel E) (28). Microscopic analysis of immunohistochemically stained CNS sections from GtQ mice infected with M-F1 CWD revealed small punctate accumulations of disease-associated CerPrP set against a background of diffuse staining (Figure 5, panel A). We observed a similar pattern in brain sections of TgE mice infected with M-F1 (Figure 5, panel B). We also detected small punctate accumulations in GtQ mice infected with M-NO1 (Figure 5, panel C). By contrast, and consistent with previous findings (27,28), GtQ mice infected with North America CWD contained intensely staining, amorphous aggregates of disease-associated CerPrP (Figure 5, panel D).
Our previous studies showed that although Gt and Tg mice expressing either CerPrPC-E226 or CerPrPC-Q226 were susceptible to North America CWD, times to disease onset were more rapid in mice expressing CerPrPC-E226 (24,26–28). By comparison, although efficient transmission of Norway moose CWD prions was registered in Gt and Tg mice expressing CerPrPC-Q226, their counterparts expressing CerPrPC-E226 were resistant to infection or had prolonged incubation times and incomplete attack rates (28). Those studies also revealed strain variation among Norway moose CWD isolates, the properties of which were different from Norway reindeer CWD (28). The different properties of emergent Norway strains compared with North America CWD made it unlikely that they were causally related. Parallel experiments in bank voles supported this interpretation (35). Differences in the strain properties of M-F1 and North America CWD enable us to build on our previous conclusions (28,35) and to state more broadly that the etiology of Nordic CWD is distinct from North America CWD.
Our analyses reveal that certain characteristics of M-F1 CWD prions overlap with those of Norway moose CWD. M-F1 and Norway moose CWD both propagated more efficiently in mice expressing CerPrPC-Q226. Western and histoblot profiles of M-F1 and M-NO1 CerPrPSc are also indistinguishable. Our findings in this study show that M-F1 prions, like Norway moose CWD prions, are nonlymphotropic. Despite these broadly similar characteristics, other measures, including consistently different disease kinetics, distinct neuropathologic lesion profiles, and variable responses to GdnHCl denaturation, indicate that the strain properties of Finland and Norway moose CWD prions are different. This finding adds to an increasing body of evidence for a surprising variety of strains among Nordic cervids, which stands in contrast to the relatively consistent CWD strain profile among North America deer, elk, and moose. As yet, there appears to be no clear explanation for this diversity of Nordic CWD strains or insights into their origins. Although efficient prion dissemination to peripheral tissues of affected cervids has been cited as a reason for the contagious properties of North America CWD (36), the nonlymphotropic properties of Finland and Norway moose CWD strains suggests a decreased potential for contagious propagation. The emergence of increasing numbers of CWD strains in a confined geographic location during a short timeframe presents challenges when considering a sporadic etiology; however, our findings showing dissimilar CWD profiles in Nordic moose can be explained by divergent strains arising from unrelated, stochastic conversion events in different animals. CWD has also been diagnosed in moose in Sweden (23). Analyses of the strain properties of those additional emergent CWD cases will be of considerable interest.
Additional findings from this study build on our previous observations and warrant discussion. Seminal studies indicating that related PrPSc and PrPC primary structures supported optimal prion propagation laid the foundation for the development of Tg mice with susceptibility to human and animal prions (37–39). Because moose express CerPrPC-Q226, we reasoned that the relatively inefficient transmission of M-F1 prions in GtE and TgE mice might be overcome by acclimatization in mice expressing CerPrPC-E226, resulting in adapted M-F1 prions composed of CerPrPSc-E226. However, our findings showing comparable transmission profiles of TgE (M-F1) and TgQ (M-F1) prions indicate that M-F1 favors conformational conversion of CerPrPC-Q226 over CerPrPC-E226 regardless of whether prions are composed of CerPrPSc-E226 or CerPrPSc-Q226. Resistance of M-F1 prions to adapt to a host expressing a single amino acid difference is reminiscent of our description of nonadaptive prion amplification (NAPA) (40). Those studies showed that prions produced during particular interspecies transmissions are not adapted for sustained propagation in that new host. Instead, prions resulting from NAPA retain atavistic preferences for their species of origin (40). In this context, although M-F1 prions are capable of suboptimal propagation by NAPA using CerPrPC-E226 as template for replication, the resulting TgE (M-F1) prions are not adapted for further propagation by CerPrPC-E226 but instead retain a preference for conversion of CerPrPC-Q226.
Ms. Sun is a PhD candidate in the Cell and Molecular Biology Program at Colorado State University. Her primary research interests are focused on characterizing novel strains of chronic wasting disease and ascertaining the mechanism by which prions encipher strain properties.
Acknowledgment
We thank Marja Isomursu for CWD surveillance and sample collection in Finland.
References
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP(121-231). Nature. 1996;382:180–2. DOIPubMedGoogle Scholar
- Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, et al. High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell. 2021;81:4540–4551.e6. DOIPubMedGoogle Scholar
- Kang HE, Bian J, Kane SJ, Kim S, Selwyn V, Crowell J, et al. Incomplete glycosylation during prion infection unmasks a prion protein epitope that facilitates prion detection and strain discrimination. J Biol Chem. 2020;295:10420–33. DOIPubMedGoogle Scholar
- Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46:417–28. DOIPubMedGoogle Scholar
- Hoyt F, Standke HG, Artikis E, Schwartz CL, Hansen B, Li K, et al. Cryo-EM structure of anchorless RML prion reveals variations in shared motifs between distinct strains. Nat Commun. 2022;13:4005. DOIPubMedGoogle Scholar
- Manka SW, Zhang W, Wenborn A, Betts J, Joiner S, Saibil HR, et al. 2.7 Å cryo-EM structure of ex vivo RML prion fibrils. Nat Commun. 2022;13:4004. DOIPubMedGoogle Scholar
- Telling GC. The shape of things to come: structural insights into how prion proteins encipher heritable information. Nat Commun. 2022;13:4003. DOIPubMedGoogle Scholar
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. DOIPubMedGoogle Scholar
- Büeler H, Aguzzi A, Sailer A, Greiner R-A, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–47. DOIPubMedGoogle Scholar
- Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. DOIPubMedGoogle Scholar
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science. 2004;305:673–6. DOIPubMedGoogle Scholar
- Bartz JC. Prion strain diversity. Cold Spring Harb Perspect Med. 2016;6:
a024349 . DOIPubMedGoogle Scholar - Will RG, Cousens SN, Farrington CP, Smith PG, Knight RSG, Ironside JW. Deaths from variant Creutzfeldt-Jakob disease. Lancet. 1999;353:979. DOIPubMedGoogle Scholar
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–82. DOIPubMedGoogle Scholar
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–68. DOIPubMedGoogle Scholar
- Benestad SL, Telling GC. Chronic wasting disease: an evolving prion disease of cervids. Handb Clin Neurol. 2018;153:135–51. DOIPubMedGoogle Scholar
- Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis. 1980;16:89–98. DOIPubMedGoogle Scholar
- US Geological Survey. Expanding distribution of chronic wasting disease. 2022 Sep 26 [cited 2022 Dec 22]. https://www.usgs.gov/centers/nwhc/science/expanding-distribution-chronic-wasting-disease
- Sohn HJ, Kim JH, Choi KS, Nah JJ, Joo YS, Jean YH, et al. A case of chronic wasting disease in an elk imported to Korea from Canada. J Vet Med Sci. 2002;64:855–8. DOIPubMedGoogle Scholar
- Kim TY, Shon HJ, Joo YS, Mun UK, Kang KS, Lee YS. Additional cases of Chronic Wasting Disease in imported deer in Korea. J Vet Med Sci. 2005;67:753–9. DOIPubMedGoogle Scholar
- Lee YH, Sohn HJ, Kim MJ, Kim HJ, Park KJ, Lee WY, et al. Experimental chronic wasting disease in wild type VM mice. J Vet Med Sci. 2013;75:1107–10. DOIPubMedGoogle Scholar
- Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikøren T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet Res (Faisalabad). 2016;47:88. DOIPubMedGoogle Scholar
- Tranulis MA, Gavier-Widén D, Våge J, Nöremark M, Korpenfelt SL, Hautaniemi M, et al. Chronic wasting disease in Europe: new strains on the horizon. Acta Vet Scand. 2021;63:48. DOIPubMedGoogle Scholar
- Angers RC, Seward TS, Napier D, Green M, Hoover E, Spraker T, et al. Chronic wasting disease prions in elk antler velvet. Emerg Infect Dis. 2009;15:696–703. DOIPubMedGoogle Scholar
- Angers RC, Kang HE, Napier D, Browning S, Seward T, Mathiason C, et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science. 2010;328:1154–8. DOIPubMedGoogle Scholar
- Angers R, Christiansen J, Nalls AV, Kang HE, Hunter N, Hoover E, et al. Structural effects of PrP polymorphisms on intra- and interspecies prion transmission. Proc Natl Acad Sci U S A. 2014;111:11169–74. DOIPubMedGoogle Scholar
- Bian J, Christiansen JR, Moreno JA, Kane SJ, Khaychuk V, Gallegos J, et al. Primary structural differences at residue 226 of deer and elk PrP dictate selection of distinct CWD prion strains in gene-targeted mice. Proc Natl Acad Sci U S A. 2019;116:12478–87. DOIPubMedGoogle Scholar
- Bian J, Kim S, Kane SJ, Crowell J, Sun JL, Christiansen J, et al. Adaptive selection of a prion strain conformer corresponding to established North American CWD during propagation of novel emergent Norwegian strains in mice expressing elk or deer prion protein. PLoS Pathog. 2021;17:
e1009748 . DOIPubMedGoogle Scholar - Browning SR, Mason GL, Seward T, Green M, Eliason GA, Mathiason C, et al. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol. 2004;78:13345–50. DOIPubMedGoogle Scholar
- Kang HE, Weng CC, Saijo E, Saylor V, Bian J, Kim S, et al. Characterization of conformation-dependent prion protein epitopes. J Biol Chem. 2012;287:37219–32. DOIPubMedGoogle Scholar
- Taraboulos A, Jendroska K, Serban D, Yang S-L, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brain. Proc Natl Acad Sci U S A. 1992;89:7620–4. DOIPubMedGoogle Scholar
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–11. DOIPubMedGoogle Scholar
- Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron. 2002;34:921–32. DOIPubMedGoogle Scholar
- Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005;4:805–14. DOIPubMedGoogle Scholar
- Nonno R, Di Bari MA, Pirisinu L, D’Agostino C, Vanni I, Chiappini B, et al. Studies in bank voles reveal strain differences between chronic wasting disease prions from Norway and North America. Proc Natl Acad Sci U S A. 2020;117:31417–26. DOIPubMedGoogle Scholar
- Davenport KA, Christiansen JR, Bian J, Young M, Gallegos J, Kim S, et al. Comparative analysis of prions in nervous and lymphoid tissues of chronic wasting disease-infected cervids. J Gen Virol. 2018;99:753–8. DOIPubMedGoogle Scholar
- Prusiner SB, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–86. DOIPubMedGoogle Scholar
- Scott M, Groth D, Foster D, Torchia M, Yang S-L, DeArmond SJ, et al. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell. 1993;73:979–88. DOIPubMedGoogle Scholar
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. DOIPubMedGoogle Scholar
- Bian J, Khaychuk V, Angers RC, Fernández-Borges N, Vidal E, Meyerett-Reid C, et al. Prion replication without host adaptation during interspecies transmissions. Proc Natl Acad Sci U S A. 2017;114:1141–6. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: January 13, 2023
Table of Contents – Volume 29, Number 2—February 2023
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Glenn Telling, Colorado State University, 300 W Lake St, Fort Collins, CO 80523, USA
Top