Volume 32, Number 3—March 2026
Research
Strongyloides Genetic Diversity among Humans, Dogs, and Nonhuman Primates, Central African Republic, 2016–2022
Cite This Article
Citation for Media
Abstract
Strongyloides stercoralis nematode infection occurs in ≈600 million persons worldwide and is listed by the World Health Organization as a neglected tropical disease. Understanding zoonotic potential is critical, especially in areas where humans, domestic animals, and wildlife interact. We explored cross-species sharing of Strongyloides nematodes by analyzing fecal samples from humans, dogs, and nonhuman primates in the Dzanga-Sangha Protected Areas, Central African Republic. We detected positive samples by quantitative PCR and assessed genetic diversity through amplification of the 18S rRNA HVR-IV region and cox1, followed by high-throughput sequencing. Strongyloides prevalence was high in humans, dogs, and gorillas. S. stercoralis haplotype A nematode dominated in humans but appeared in dogs and apes, whereas S. fuelleborni nematode was present in all hosts. Shared species and haplotypes indicated zoonotic transmission. Our findings highlight the need for molecular surveillance and emphasize the role of dogs and nonhuman primates as reservoirs, complicating efforts to control infections in human populations.
Strongyloidiasis, caused by Strongyloides nematodes, is a zoonotic disease with public health and veterinary implications worldwide (1). The World Health Organization classifies strongyloidiasis among neglected tropical diseases requiring urgent control in endemic regions (2). Current estimates suggest that >600 million persons are infected globally, predominantly in tropical and subtropical areas (3). Strongyloides stercoralis and S. fuelleborni nematodes are the main species infecting humans; transmission typically occurs through transcutaneous exposure. More severe infections are primarily attributed to S. stercoralis nematodes, which is capable of autoinfection and can lead to severe systemic disease, particularly in immunocompromised persons; infection can result in death in extreme cases. Uncomplicated infections often manifest in gastrointestinal, pulmonary, or dermatological symptoms (4).
Molecular analyses of S. stercoralis nematodes have revealed 2 primary lineages: the potentially zoonotic lineage A, which involves dogs as potential reservoirs for human infection, and lineage B, which is largely restricted to canine hosts (5,6). Conversely, S. fuelleborni nematodes, although less common, are confined to nonhuman primates (NHPs) in Africa and Asia, occasionally spilling over to humans (1). Despite numerous documented cases, the true global prevalence of strongyloidiasis remains uncertain because of limited surveillance and underdiagnosis, compounded by the asymptomatic nature of many infections and the lack of standardized diagnostic tools (7,8).
The Central African Republic (CAR), one of the world’s most resource-constrained countries (9), ranks 191st out of 193 on the 2022 United Nations Human Development Index (10). Its tropical ecosystems, which are rich in biodiversity and wildlife (11), create favorable conditions for zoonotic disease circulation (12). Frequent interactions among humans, NHPs, domestic animals, and wildlife encourage the spread of infectious diseases (13), including Strongyloides nematodes (14). Dogs often act as ecologic bridges as a result of their hunting and scavenging behaviors (15). Soil-transmitted helminth infections remain among the most prevalent public health challenges in CAR (16); Strongyloides nematodes pose a particular concern for both human and NHP populations (14,17).
This study investigates the role of potential animal reservoirs in human Strongyloides infections within the Dzanga-Sangha Protected Areas (DSPA), CAR, where humans, NHPs, and domestic dogs interact closely at the human–wildlife interface. By using molecular genotyping, we analyzed the diversity and sharing of Strongyloides haplotypes among humans, NHPs, and dogs to inform control strategies in complex multihost systems.
Study Design and Participants
Figure 1

Figure 1. Location of study site in analysis of Strongyloidesgenetic diversity among humans, dogs, and nonhuman primates, Dzanga-Sangha Protected Areas, Central African Republic, 2016–2022. Inset shows location of Central African...
The study was conducted in the DSPA in CAR (Figure 1) during 2016–2022, a location known for habituation of western lowland gorillas (Gorilla gorilla gorilla) to human presence and home of traditional BaAka hunter-gatherer communities living in close contact with wildlife. DSPA consists of several zones at varying levels of protection, including the strictly protected Dzanga-Ndoki National Park and the Dzanga-Sangha Special Reserve, a multiuse area where human activities are regulated to varying extents (Figure 1). The research complied with the legal requirements of the CAR and with all research, ethical, and sample transport approvals (Appendix 1).
Samples were collected from humans and domestic dogs from 2 villages located within the Special Reserve, whereas wildlife samples were collected in the National Park. Fresh stool samples were obtained from BaAka trackers who worked directly with NHPs in the National Park and also entered the Special Reserve (n = 18), as well as from humans residing in the villages (n = 32), who access the Special Reserve for daily activities such as gathering or hunting. Samples were also collected from dogs (n = 47), which were further categorized as hunting dogs entering the Special Reserve (n = 35) and guarding dogs (n = 12). Wildlife samples consisted of samples from western lowland gorillas (n = 101) at different levels of habituation, unhabituated central chimpanzees (Pan troglodytes troglodytes, n = 7), and a habituated group of agile mangabeys (Cercocebus agilis, n = 50). Samples were collected as part of health surveillance efforts (Appendix 2 Table 1). Approximately 1 g of feces was collected and immediately fixed in 96% ethanol and stored at –20°C before DNA isolation.
Procedures
We dried fecal samples overnight at 37°C to evaporate the ethanol. We isolated total DNA using the PowerSoil DNA isolation kit (QIAGEN, https://www.qiagen.com). The extracted DNA was first screened by quantitative PCR (qPCR) targeting the small subunit (18S) rRNA gene specific to the genus Strongyloides (19) using the LightCycler 480 Real-Time PCR system (Roche, https://www.roche.com). We only included samples positive for Strongyloides in high-throughput library preparation. We used DNA extractions from Strongyloides PCR-negative feces and water as negative controls, whereas S. stercoralis first-stage larva was the positive control. All samples were prepared in technical PCR replicates (20). We prepared the high-throughput sequencing library using a 3-step PCR approach (Appendix 2 Table 2). We designed primers for the first PCR step, nested for the hypervariable region IV (HVR-IV) of the 18S rRNA gene and seminested for a portion of the mitochondrial cytochrome c oxidase subunit 1 gene (cox1), to increase sensitivity and ensure sufficient yield of amplicons for downstream sequencing (Appendix 1). In the second PCR step, we amplified the HVR-IV-18S rRNA and cox1 (21). In the final PCR step, we applied Nextera primers with unique barcodes for each technical PCR replicate. We performed paired-end sequencing (2 × 300 bp) on the MGI DNBSEQ-G400 platform (MGI Tech, https://mgi-tech.eu).
We first demultiplexed raw fastq sequences and trimmed primer sequences using skewer version 0.2.2 (22). We then filtered, dereplicated, and denoised trimmed sequences and merged paired reads in R version 4.2.2 using the dada2 package (23). After processing, the final amplicon lengths were 255 bp for the 18S rRNA region and 217 bp for the cox1. During merging, we marked amplicon sequencing variants (ASVs) inconsistently present in both PCR technical replicates as potential artifacts and removed them from downstream analyses. We searched for corresponding sequences against the National Center for Biotechnology Information Nucleotide database (downloaded in February 2024) and excluded environmental, uncultured, <85% identity, and <90% coverage hits. We downloaded taxonomy using taxonomizer and used the created reference database to assign a taxonomic classification in our dataset through dada2’s Assign Taxonomy method (24).
Statistical Analysis
We analyzed all data using the statistical software RStudio (https://www.rstudio.com). We assessed statistical differences in Strongyloides occurrence between trackers and villagers and between hunting and guard dogs using the χ2 test. We generated a bar plot to depict the proportion of Strongyloides haplotypes of HVR-IV-18S rRNA in each sample for better resolution. For cox1 haplotypes, we constructed a median-joining network and visualized using Population Analysis with Reticulate Trees (PopART, https://popart.maths.otago.ac.nz). In addition, we performed a phylogenetic analysis using the MrBayes plugin in Geneious 9.1.5 (https://www.geneious.com) to complement the network approach and assess the phylogenetic placement of the detected haplotypes. To examine differences in α diversity, assessed as ASVs richness (number of ASVs per sample), we applied a generalized linear model (GLM) with a quasipoisson error distribution. We conducted posthoc pairwise comparisons using the Tukey test to identify specific differences among the studied host groups. We evaluated community composition diversity by analyzing the relative representation of HVR-IV-18S rRNA and cox1 ASVs using Bray-Curtis ecologic distances. We visualized the clustering patterns with principal coordinate analysis (PCoA). To test for interspecific differences in Strongyloides nematode community composition among hosts, we performed a permutational analysis of variance (PERMANOVA), followed by similarity analysis (ANOSIM).
Strongyloides qPCR Detection
The number of Strongyloides-positive samples by qPCR was high across all studied host species; rates were 76% (38/50) in humans, 60% (28/47) in dogs, 59% (58/101) in gorillas, 43% (3/7) in chimpanzees, and 38% (19/50) in mangabeys. Among humans, BaAka gorilla trackers exhibited a slightly higher, although nonsignificant (χ2 = 0.83; p = 0.36), prevalence of infection (83.3% [15/18]) than that observed in villagers (71.9% [23/32]). Similarly, hunting dogs, which actively venture into the forest, showed a significantly (χ2 = 12.32; p = 0.0004) higher prevalence (74.3% [26/35]) than did guard dogs, which remain in villages (16.7% [2/12]). All qPCR-positive samples were further successfully sequenced with both targets (HVR-IV-18S rRNA and cox1), except for 3 mangabey samples that were near the detection threshold (cycle threshold ≈33–34), which likely explains their sequencing failure.
Strongyloides HVR-IV-18S rRNA Diversity
Genotyping identified a total of 11,472,181 high-quality HVR-IV-18S rRNA reads, with a median sequencing depth of 74,200 (range 1,221–302,518) per sample. Taxonomic analysis identified a total of 24 ASVs (Table). Six clustered into already known Strongyloides haplotypes: haplotype A of S. stercoralis and haplotypes K, L, M, P, and T of S. fuelleborni. Five S. fuelleborni ASVs did not match with any previously described haplotypes. Those ASVs were detected in only 9 samples (9/140 [6.4%]). Three haplotypes were host-specific (detected exclusively in human, dog, or gorilla), whereas 2 haplotypes were shared (1 between gorilla and mangabey and the other between human and dog). Six ASVs were classified as uncharacterized Strongyloides species and were found in 14 samples (14/140 [10%]). Three ASVs were detected in a single host, each occurring exclusively in mangabeys, whereas the remaining 3 were shared (2 between humans and dogs, and 1 within mangabeys). Last, 7 unassigned ASVs, detected in 19% of the samples, were tentatively classified as being most closely related to the order Rhabditida.
Figure 2
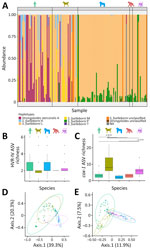
Figure 2. Relative community composition of Strongyloides HVR-IV-18S rRNA haplotypes across examined hosts in study of Strongyloidesgenetic diversity among humans, dogs, and nonhuman primates, Dzanga-Sangha Protected Areas, Central...
Overall, S. fuelleborni nematodes dominated across all studied hosts; haplotype L was the most prevalent variant (93.8% of total prevalence). Potentially zoonotic haplotype A of S. stercoralis was detected in 29% of samples from humans, dogs, and gorillas (Table). Relative abundances bar plot of Strongyloides HVR-IV-18S rRNA ASVs shows interspecific differences in Strongyloides community composition depending on host species (Figure 2). Humans were predominantly infected with S. fuelleborni haplotype L and S. stercoralis haplotype A (Table; Figure 2, panel A). S. stercoralis haplotype A was detected less frequently in BaAka gorilla trackers (67%) than in their village-dwelling relatives (78%), although that difference was not statistically significant (χ2 = 0.63; p = 0.43); haplotype A was absent in guard dogs but present in 30% of hunting dogs. NHPs were mainly infected with S. fuelleborni haplotype L. Unclassified S. fuelleborni and other unassigned Strongyloides variants were predominantly found in mangabeys (Table).
Zoonotic Potential of Strongyloides Nematode on the Basis of HVR-IV-18S rRNA
The highest number of Strongyloides HVR-IV-18S rRNA ASVs (8) was shared between humans and dogs, accounting for 33.3% of all observed ASVs. Those included haplotype A of S. stercoralis, haplotypes K, L, M, and P of S. fuelleborni, 1 unclassified S. fuelleborni (ASV_19), and 2 unidentified Strongyloides spp. (ASV_14 and ASV_21). Four ASVs (16.6% of all observed ASVs), haplotype A of S. stercoralis, and haplotypes K, L, and P of S. fuelleborni were shared between humans and NHPs. Haplotype A of S. stercoralis and haplotypes K, L, and P of S. fuelleborni were shared across humans, dogs, and gorillas (Figure 2, panel A).
Differences in Strongyloides HVR-IV-18S rRNA Communities
The number of ASVs per sample did not differ significantly among host species, as determined by a GLM (p>0.1) and subsequent Tukey test (Figure 2, panel B). However, the PCoA diagram based on Bray-Curtis ecologic distances revealed distinct differences in Strongyloides community composition among humans, dogs, and NHPs (Figure 2, panel D). Those differences were further statistically confirmed by PERMANOVA (F(4,137) = 11.372; p = 0.001) and ANOSIM (R = 0.3435; p = 0.001) tests. The overlap in Strongyloides communities was greater between humans and dogs, whereas communities within NHPs were more similar but distinct from those of humans and dogs (Figure 2, panel D).
Strongyloides cox1 Diversity and Zoonotic Potential
Figure 3

Figure 3. Median-joining Strongyloides haplotype network for the mitochondrial cox1 gene studied in various host species in study of Strongyloidesgenetic diversity among humans, dogs, and nonhuman...
Figure 4

Figure 4. Bayesian phylogenetic tree based on Strongyloides spp. cox1 (700 bp) sequences derived from study of Strongyloidesgenetic diversity among humans, dogs, and nonhuman primates, Dzanga-Sangha...
We identified a total of 3,381,999 reads based on cox1; the median sequencing depth was 2,314 (range 75–56,108) per sample. Taxonomic assignment revealed 62 Strongyloides ASVs and 72 unassigned variants, which we tentatively classified as closest to the order Rhabditida. The 62 Strongyloides ASVs were used to construct a median-joining phylogenetic network (Figure 3) and Bayesian phylogeny (Figure 4).
The nucleotide diversity was high (N = 0.076), and 52 of the ASVs were parsimony informative. The haplotype network consisted of 25 S. stercoralis, 31 S. fuelleborni, and 6 unclassified Strongyloides ASVs. The network revealed divergence in ASVs on the basis of host specificity. S. stercoralis ASVs were primarily shared between humans and dogs, with occasional overlap with gorillas. Haplotype ASV_7 was most common in dogs, whereas ASV_14 was most frequent in humans. Chimpanzees shared only 1 haplotype (ASV_117) with gorillas, and intriguingly, haplotype ASV_43 was found exclusively in gorillas (Figure 3). In the phylogenetic tree, all ASVs of S. stercoralis cluster within potentially zoonotic lineage A (Figure 4). S. fuelleborni ASVs all clustered to S. fuelleborni Africa clade (Figure 4) and displayed host-specific divergence (Figure 3). Humans shared S. fuelleborni haplotypes mostly with dogs and occasionally with gorillas, whereas other haplotypes were restricted to NHPs. The ASV_2 haplotype was most common in both humans and dogs, whereas ASV_6 was predominant in gorillas and mangabeys. Haplotypes of unclassified Strongyloides species were detected only in dogs (Figure 4). The first cluster, consisting of ASV_30, ASV_99, and ASV_191, formed a well-supported branch closely related to S. venezuelensis. The second cluster, represented by ASV_183 and ASV_72, grouped between S. planiceps and S. ratti but with low nodal support. The final lineage, ASV_102, formed a separate, also weakly supported branch positioned between S. ratti, S. procyonis, and unclassified Strongyloides spp. found in Bornean slow lorises. Overall, the median-joining phylogenetic network and Bayesian phylogeny were highly complementary, revealing host-associated divergence in S. stercoralis and S. fuelleborni and highlighting distinct clustering of unclassified Strongyloides taxa.
Differences in Strongyloides cox1 Communities
cox1 ASV richness differed significantly among the studied hosts (GLM F(4,134) = 253.93; p<0.0001). Tukey posthoc testing revealed that dogs differed significantly from all other hosts (p<0.01 for all pairwise comparisons), whereas mangabeys also differed significantly from humans and gorillas (p<0.01) (Figure 2, panel C). We observed no significant differences between the remaining host pairs. The PCoA diagram based on Bray-Curtis ecologic distances further confirmed clear differences between humans, dogs, and NHPs (Figure 2, panel E), supporting the findings of the haplotype network analysis. Those differences in the composition of Strongyloides infections between host species were further statistically confirmed by PERMANOVA (F(4,135) = 11.192; p = 0.001) and ANOSIM (R = 0.3438; p = 0.001) tests.
We investigated Strongyloides nematodes diversity and haplotype sharing among humans, dogs, and NHPs cohabiting in the DSPA, CAR, to assess zoonotic potential and multihost transmission (25). A previous study from the DSPA investigated Strongyloides nematodes in humans, gorillas, and chimpanzees, reporting S. stercoralis exclusively in humans and identifying different haplotypes of S. fuelleborni in gorillas and chimpanzees than those found in humans (14). However, that study was limited by a small sample size of larvae and did not include dogs, which are potential hosts. Our study highlights the critical importance of employing modern diagnostic approaches and examining a broader range of hosts from the same locality to better understand Strongyloides sharing in the ecosystem.
Our genotyping approach revealed a remarkable degree of genetic diversity within Strongyloides species, reflecting the complexity of their populations, and further identified the sharing of specific haplotypes across different host species. Of note, S. fuelleborni, likely originating from NHPs, dominated across all hosts; haplotype L was the most prevalent variant. Zoonotic S. stercoralis haplotype A was observed primarily in humans, dogs, and gorillas. This variation in prevalence underscores the host-specific transmission dynamics and the role of local ecologic factors in determining Strongyloides distribution. Those findings emphasize the need for a One Health approach (26) because of the zoonotic potential of S. stercoralis nematodes and the species’ strong connection to the environment.
Despite the identification of dominant haplotypes, the proportion of Strongyloides sequences remains unclassified, highlighting the limitations of short-marker–based phylogenetic inference. Unclassified S. fuelleborni haplotypes were detected in all host species except chimpanzees, for which only a limited dataset was available. Those sequences likely reflect previously undetected diversity, because additional haplotypes have been reported with expanded sampling. Given the limited data currently available from sites in Africa (27), further haplotypes are expected to emerge as sampling across hosts and regions improves.
Several unclassified ASVs clustered with S. venezuelensis with high nodal support, whereas other unclassified ASVs formed clusters with very low support, rendering their classification unreliable. The placement of such lineages, based solely on short cox1 fragments, is highly challenging, because those sequences might lack sufficient phylogenetically informative sites to resolve relationships among closely related or cryptic taxa. Similar issues have been reported previously (e.g., short cox1 fragments were insufficient for precise taxonomic resolution of cryptic Strongyloides in dogs [28]). Therefore, integrating comprehensive genetic data, such as longer mitochondrial sequences or genomic information from individual larvae, with morphological analyses of larvae and adult or paratenic females is crucial for resolving the taxonomy and evolutionary relationships of unclassified Strongyloides spp.
We recognize the inherent limitations of single-locus genotyping, because reliance on a single marker provides a restricted representation of genetic diversity and might not reflect genome-wide variation. That limitation means substantially larger host sample sizes are required to achieve the same statistical power and robustness that whole-genome data can provide, because only by averaging across many individuals can stochastical effects at a single locus be mitigated. We also acknowledge that our reliance on fecal DNA, although advantageous as a noninvasive sampling method, can introduce issues related to DNA degradation, contamination, and allelic dropout, further constraining data quality. Thus, our conclusions should be interpreted with caution and viewed as complementary to, rather than a substitute for, genome-wide analyses.
Strongyloides communities were more similar between humans and dogs than between either of those hosts and NHPs. The highest proportion of shared haplotypes was observed between humans and dogs, accounting for 33.3% of all detected ASVs. This pattern supports the involvement of dogs in the circulation of S. stercoralis nematodes alongside humans in the DSPA (5,6). Consistent with this pattern, S. stercoralis haplotype A predominated in humans, suggesting that human-associated circulation represents a key component of Strongyloides nematodes occurrence in this setting and that dogs potentially act as secondary hosts.
Unexpectedly, sharing between humans and dogs was also observed for S. fuelleborni haplotypes K and M, a species previously considered to circulate primarily in NHPs (14). The presence of haplotype M in both humans and dogs, with higher prevalence in dogs, is notable given its limited geographic reporting to date in humans, restricted to Senegal (21) and CAR (14). The high prevalence in dogs suggests a potential contribution to the environmental dissemination of S. fuelleborni nematodes within DSPA, although spurious infections associated with coprophagy cannot be excluded (29). Experimental infections of dogs with S. fuelleborni nematodes have been demonstrated (30), but natural infections remain unconfirmed, and fecal metabarcoding alone cannot resolve the definitive host status of dogs.
ASVs shared between humans and NHPs included S. stercoralis haplotype A and S. fuelleborni haplotypes K, L, and P, accounting for 16.6% of all observed ASVs; all of those were also detected in dogs. Strongyloides fuelleborni haplotype L, which is well established across all studied hosts, was previously reported only in gorillas (14) and in African vervets on St. Kitts (31). Although its origin remains speculative, its presence across host species suggests extensive host sharing within the DSPA. In contrast, haplotype P was predominantly detected in NHPs, which likely represent a key source of exposure for humans and dogs. Together, these patterns indicate shared circulation of Strongyloides nematodes among humans, dogs, and NHPs in the DSPA.
Given the soil-transmitted nature of Strongyloides nematodes and the close coexistence of hosts in DSPA, environmental exposure represents a major challenge for disease control (4). Although chemotherapy is effective in the short term to treat strongyloidiasis (32), high reinfection rates, autoinfection, free-living stages in soil, multiple host species, and emerging concerns about drug resistance complicate long-term elimination, particularly in tropical settings (33,34), highlighting the need for a comprehensive strategy. A crucial aspect of controlling strongyloidiasis is recognizing the role of wildlife in transmission. Because treating wildlife such as free-ranging NHPs is not feasible, effective control requires a One Health approach that considers humans, domestic animals, and the environment (26). Practical measures, including proper latrine use and use of footwear to reduce soil contact, should complement treatment of infected persons (35). Ivermectin remains the treatment of choice (33), but its use must be carefully considered in regions with high Loa loa microfilaremia prevalence (36), as in CAR. Overall, the potential for rapid reinfection from multiple hosts underscores the need for integrated control strategies.
In conclusion, the close coexistence of multiple host species sharing Strongyloides infections in the DSPA likely contributes to the persistence of the parasite in this environment. Expanding investigations beyond humans to include domestic animals and wildlife is therefore essential, and our findings highlight the relevance of a One Health approach that integrates those host groups (37). The observed overlap of S. stercoralis and S. fuelleborni haplotypes among humans, dogs, and NHPs reflects the complexity of host associations within this ecosystem. By adopting a multihost diagnostic approach, we provide direct insights into Strongyloides nematodes sharing among hosts in the DSPA, informing more realistic and sustainable control strategies.
Dr. Nosková is a parasitologist specializing in zoonotic nematodes and parasite transmission between humans, domestic animals, and wildlife. Her research focuses on the molecular epidemiology, host specificity, and phylogeny of Strongyloides spp., with additional experience in veterinary parasitology, paleoparasitology, and wildlife conservation.
Acknowledgments
We would like to express our gratitude to the government of the Central African Republic and the World Wildlife Fund for granting permission to conduct our research; the Ministre de l’Enseignement Supérieur, de la Recherche Scientifique et de l’Innovation Technologique, for providing research permits; and the Primate Habituation Programme for providing logistical support in the field. Finally, yet importantly, we would like to thank all the trackers and assistants in Dzanga-Sangha Protected Areas.
Raw HVR-IV-18S rRNA and cox1 sequencing data are archived in the European Nucleotide Archive and are available project accession no. PRJEB101217. Accession numbers for each samples are available in Appendix 2 Table 3.
This project was supported by Czech Science Foundation 22-16475S and partially supported by Masaryk University internal grant no. MUNI/A/1602/2023. We acknowledge the CF Genomics supported by the NCMG research infrastructure (LM2023067 funded by MEYS CR) for their support with obtaining scientific data presented in this paper. Computational resources were provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.
References
- Bradbury RS, Pafčo B, Nosková E, Hasegawa H. Strongyloides genotyping: a review of methods and application in public health and population genetics. Int J Parasitol. 2021;51:1153–66. DOIPubMedGoogle Scholar
- Asundi A, Beliavsky A, Liu XJ, Akaberi A, Schwarzer G, Bisoffi Z, et al. Prevalence of strongyloidiasis and schistosomiasis among migrants: a systematic review and meta-analysis. Lancet Glob Health. 2019;7:e236–48. DOIPubMedGoogle Scholar
- Buonfrate D, Bisanzio D, Giorli G, Odermatt P, Fürst T, Greenaway C, et al. The global prevalence of Strongyloides stercoralis infection. Pathogens. 2020;9:468. DOIPubMedGoogle Scholar
- Nutman TB. Human infection with Strongyloides stercoralis and other related Strongyloides species. Parasitology. 2017;144:263–73. DOIPubMedGoogle Scholar
- Jaleta TG, Zhou S, Bemm FM, Schär F, Khieu V, Muth S, et al. Different but overlapping populations of Strongyloides stercoralis in dogs and humans-dogs as a possible source for zoonotic strongyloidiasis. PLoS Negl Trop Dis. 2017;11:
e0005752 . DOIPubMedGoogle Scholar - Nagayasu E, Aung MPPTHH, Hortiwakul T, Hino A, Tanaka T, Higashiarakawa M, et al. A possible origin population of pathogenic intestinal nematodes, Strongyloides stercoralis, unveiled by molecular phylogeny. Sci Rep. 2017;7:4844. DOIPubMedGoogle Scholar
- Gordon CA, Utzinger J, Muhi S, Becker SL, Keiser J, Khieu V, et al. Strongyloidiasis. Nat Rev Dis Primers. 2024;10:6. DOIPubMedGoogle Scholar
- Buonfrate D, Requena-Mendez A, Angheben A, Cinquini M, Cruciani M, Fittipaldo A, et al. Accuracy of molecular biology techniques for the diagnosis of Strongyloides stercoralis infection-A systematic review and meta-analysis. PLoS Negl Trop Dis. 2018;12:
e0006229 . DOIPubMedGoogle Scholar - Kuehne A, Roberts L. Learning from health information challenges in the Central African Republic: where documenting health and humanitarian needs requires fresh approaches. Confl Health. 2021;15:68. DOIPubMedGoogle Scholar
- United Nations Development Programme. Human development insights [cited 2025 Feb 6]. https://hdr.undp.org/data-center/country-insights#/ranks
- Blom A, Yamindou J, Prins HHT. Status of the protected areas of the Central African Republic. Biol Conserv. 2004;118:479–87. DOIGoogle Scholar
- David PM, Nakouné E, Giles-Vernick T. Hotspot or blind spot? Historical perspectives on surveillance and response to epidemics in the Central African Republic. Int J Public Health. 2020;65:241–8. DOIPubMedGoogle Scholar
- Pedersen AB, Davies TJ. Cross-species pathogen transmission and disease emergence in primates. Ecohealth. 2009;6:496–508. DOIPubMedGoogle Scholar
- Hasegawa H, Kalousova B, McLennan MR, Modry D, Profousova-Psenkova I, Shutt-Phillips KA, et al. Strongyloides infections of humans and great apes in Dzanga-Sangha Protected Areas, Central African Republic and in degraded forest fragments in Bulindi, Uganda. Parasitol Int. 2016;65(5 Pt A):367–70. DOIPubMedGoogle Scholar
- Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–83. DOIPubMedGoogle Scholar
- World Health Organization. Soil-transmitted helminthiases [cited 2025 Feb 6]. https://www.who.int/data/gho/data/themes/topics/soil-transmitted-helminthiases.
- Pampiglione S, Ricciardi M. Parasitological survey on Pygmies in Central Africa. I. Babinga group (Central African Republic). Riv Parassitol. 1974;35:161–88.
- Hasegawa H, Modrý D, Kitagawa M, Shutt KA, Todd A, Kalousová B, et al. Humans and great apes cohabiting the forest ecosystem in central african republic harbour the same hookworms. PLoS Negl Trop Dis. 2014;8:
e2715 . DOIPubMedGoogle Scholar - Verweij JJ, Canales M, Polman K, Ziem J, Brienen EA, Polderman AM, et al. Molecular diagnosis of Strongyloides stercoralis in faecal samples using real-time PCR. Trans R Soc Trop Med Hyg. 2009;103:342–6. DOIPubMedGoogle Scholar
- Pafčo B, Čížková D, Kreisinger J, Hasegawa H, Vallo P, Shutt K, et al. Metabarcoding analysis of strongylid nematode diversity in two sympatric primate species. Sci Rep. 2018;8:5933. DOIPubMedGoogle Scholar
- Barratt JLN, Lane M, Talundzic E, Richins T, Robertson G, Formenti F, et al. A global genotyping survey of Strongyloides stercoralis and Strongyloides fuelleborni using deep amplicon sequencing. PLoS Negl Trop Dis. 2019;13:
e0007609 . DOIPubMedGoogle Scholar - Jiang H, Lei R, Ding SW, Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15:182. DOIPubMedGoogle Scholar
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. DOIPubMedGoogle Scholar
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. DOIPubMedGoogle Scholar
- Bradbury RS, Streit A. Is strongyloidiasis a zoonosis from dogs? Philos Trans R Soc Lond B Biol Sci. 2024;379:
20220445 . DOIPubMedGoogle Scholar - Lapat JJ, Opee J, Apio MC, Akello S, Ojul CL, Onekalit R, et al. A One Health approach toward the control and elimination of soil-transmitted helminthic infections in endemic areas. IJID One Health. 2024;2:100021. DOIGoogle Scholar
- Nosková E, Sambucci KM, Petrželková KJ, Červená B, Modrý D, Pafčo B. Strongyloides in non-human primates: significance for public health control. Philos Trans R Soc Lond B Biol Sci. 2024;379:
20230006 . DOIPubMedGoogle Scholar - Beknazarova M, Barratt JLN, Bradbury RS, Lane M, Whiley H, Ross K. Detection of classic and cryptic Strongyloides genotypes by deep amplicon sequencing: A preliminary survey of dog and human specimens collected from remote Australian communities. PLoS Negl Trop Dis. 2019;13:
e0007241 . DOIPubMedGoogle Scholar - Nijsse R, Mughini-Gras L, Wagenaar JA, Ploeger HW. Coprophagy in dogs interferes in the diagnosis of parasitic infections by faecal examination. Vet Parasitol. 2014;204:304–9. DOIPubMedGoogle Scholar
- Sandground JH. Biological studies on the life-cycle in the genus Strongyloides grassi, 1879. Am J Epidemiol. 1926;6:337–88. DOIGoogle Scholar
- Richins T, Sapp SGH, Ketzis JK, Willingham AL, Mukaratirwa S, Qvarnstrom Y, et al. Genetic characterization of Strongyloides fuelleborni infecting free-roaming African vervets (Chlorocebus aethiops sabaeus) on the Caribbean island of St. Kitts. Int J Parasitol Parasites Wildl. 2023;20:153–61. DOIPubMedGoogle Scholar
- Alum A, Rubino JR, Ijaz MK. The global war against intestinal parasites—should we use a holistic approach? Int J Infect Dis. 2010;14:e732–8. DOIPubMedGoogle Scholar
- Horton J. Global anthelmintic chemotherapy programs: learning from history. Trends Parasitol. 2003;19:405–9. DOIPubMedGoogle Scholar
- A F White M, Whiley H, E Ross K. A review of Strongyloides spp. environmental sources worldwide. Pathogens. 2019;8:8.PubMedGoogle Scholar
- Khieu V, Hattendorf J, Schär F, Marti H, Char MC, Muth S, et al. Strongyloides stercoralis infection and re-infection in a cohort of children in Cambodia. Parasitol Int. 2014;63:708–12. DOIPubMedGoogle Scholar
- Kamgno J, Boussinesq M, Labrousse F, Nkegoum B, Thylefors BI, Mackenzie CD. Encephalopathy after ivermectin treatment in a patient infected with Loa loa and Plasmodium spp. Am J Trop Med Hyg. 2008;78:546–51. DOIPubMedGoogle Scholar
- Zinsstag J, Kaiser-Grolimund A, Heitz-Tokpa K, Sreedharan R, Lubroth J, Caya F, et al. Advancing One human-animal-environment Health for global health security: what does the evidence say? Lancet. 2023;401:591–604. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: March 04, 2026
Table of Contents – Volume 32, Number 3—March 2026
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Barbora Pafčo, Czech Academy of Sciences, Institute of Vertebrate Biology, Květná 8, Brno 603 00, Czech Republic
Top