Volume 24, Number 10—October 2018
Research
Non-cyp51A Azole-Resistant Aspergillus fumigatus Isolates with Mutation in HMG-CoA Reductase
Cite This Article
Citation for Media
Abstract
The recent increase in azole-resistant Aspergillus fumigatus is a global concern. Identifying the mutations that confer azole resistance is essential for developing novel methods for prompt diagnosis and effective drug treatment. We screened A. fumigatus clinical isolates for novel mutations conferring azole resistance. We compared the genomic sequences of susceptible and resistant isolates without mutations in cyp51A (non-cyp51A) and found mutations in hmg1 and erg6 involved in ergosterol biosynthesis. We also found the novel mutations in these genes in azole-resistant isolates with different genetic backgrounds. The resistant isolates with mutations in hmg1 showed increased intracellular ergosterol levels compared with susceptible isolates. This finding supports the concept that the ergosterol level is a determinant for resistance to any class of azoles. Multiple isolates with increased resistance to azole possessed a mutation in hmg1, indicating that this mutation is widely present in non-cyp51A azole-resistant A. fumigatus.
Aspergillus fumigatus is known as an opportunistic pathogen of fungal infection and causes a high mortality rate among immunosuppressed patients (1,2). Azole drugs are considered the first-line therapy in various diseases of aspergillosis. The recent increase of azole drug-resistant A. fumigatus is a major limitation of therapeutic strategies in clinical settings and a concern throughout the world (3). Most azole-resistant strains harbor a mutation in the cyp51A gene, encoding a target protein for azole drugs (4). The point-mutated Cyp51A has lower binding affinity to azole drugs, causing azole resistance in these strains (5). The amino acid substitution in Cyp51A protein is thought to occur during azole therapy inside hosts. In addition, 34 bp or 46 bp of tandem repeats in the promoter region of cyp51A cause azole resistance (6). The high incidence of such tandem repeat–type resistant strains is alarming (7), and it is widely accepted that the tandem repeat–type resistant mechanism is derived from exposure to azole fungicides in the environment (8). Whereas such cyp51A-related azole-resistant strains have been reported frequently in the past 10 years, strains without any mutation in the cyp51A gene showing low susceptibility to azole drugs have, to some extent, been isolated (9). Which mutation leads to azole resistance has thus far been determined in only a few non-cyp51A azole-resistant strains. For a deeper and broader understanding of the molecular mechanisms underlying resistance to antifungal drugs, the non-cyp51A azole-resistant strains have become the focus of attention.
The cdr1B gene, encoding an ABC transporter, plays a role in azole resistance. Deletion of the gene resulted in azole-sensitive phenotypes (10), and an azole-resistant strain with constitutive expression of cdr1B has been isolated from a patient (11). The expression of cdr1B is dependent on AtrR, a recently characterized transcriptional factor (12), but the reason for the constitutive expression remains unclear. A strain showing azole resistance with a mutation (P88L) in the hapE gene has also been isolated; the mutation was discovered by genome comparison between azole-resistant strains and corresponding susceptible strains (13). A recent study showed that HapE forms a CCAAT binding complex, which plays a role in the negative regulation of cyp51A expression. The amino acid substitution in HapE (P88L) resulted in increased cyp51A expression and consequent azole resistance (14).
These studies provided convincing evidence of the roles of Cdr1B and HapE in azole adaptation in A. fumigatus. However, each of these mutations has been reported only once in clinical isolates. Furthermore, a large portion of the non-cyp51A azole-resistant A. fumigatus strains have yet to be investigated to determine which genes and mutations could be responsible for azole resistance. To fill a gap in our understanding of the mechanism for azole resistance in the non-cyp51A strains, we searched for mutations in a series of isolates derived from a single patient in Japan who had chronic pulmonary aspergillosis (CPA) and isolates from other patients with and without azole resistance.
A. fumigatus Strains
All clinical isolates used in this study were preserved at Chiba University Medical Mycology Research Center (Chiba City, Japan). We obtained 19 isolates from a patient at hospital C who had CPA (Tables 1, 2), 3 isolates (IFM 63768, IFM 63666, and IFM 63772) from another CPA patient at hospital C on different days, and additional isolates from 3 other patients with CPA at different hospitals (IFM 62140 at hospital K, IFM 64258 at hospital I, and IFM 64303 at hospital T). We also obtained 16 azole-susceptible clinical isolates (Table 3) from respiratory samples (sputa, pleural effusion, lung tissues, or bronchoalveolar fluids) or pus from a subcutaneous abscess from patients with aspergillosis in various hospitals in Japan.
Microsatellite Genotyping
For genotyping, we PCR amplified and sequenced 6 loci of ≈400 bp from all of the isolates as described previously (15). We counted the repeat numbers of each region (2A, 2B, 2C, 3A, 3B, 3C, 4A, 4B, and 4C) from the sequence.
Antifungal Susceptibility Testing
We performed antifungal susceptibility testing as described previously (16). In brief, we performed tests in triplicate using amphotericin B (AMPH), itraconazole (ITCZ), voriconazole (VRCZ), and posaconazole (PSCZ) in RPMI 1640 medium (pH 7.0) at 35°C, according to the Clinical and Laboratory Standards Institute reference method for broth microdilution (https://clsi.org/standards/products/microbiology/documents/m38), with partial modifications using the dried plate for antifungal susceptibility testing (Eiken Chemicals, Tokyo, Japan).
Sequencing cyp51A, erg6, and hmg1 Genes
To detect mutations in cyp51A, erg6, and hmg1 genes, we performed PCR amplification and sequenced these regions using appropriately designed primers (Technical Appendix Table 1). We performed sequence variant detection by comparison with reference sequences from GenBank and Fungi DB (http://fungidb.org/fungidb/) (GenBank accession no. AF338659 for cyp51A, Fungi DB accession nos. AFUB_020770 for hmg1, and AFUB_099400 for erg6).
Whole-Genome Sequencing
We performed whole-genome sequencing by using next-generation methods as described previously (15). In brief, we extracted genome DNA from overnight-cultured mycelia by DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) or by phenol-chloroform extraction and Nucleobond AXG column (TaKaRa, Kusatsu, Japan) with Nucleobond Buffer Set III (TaKaRa). For analyzing IFM 60814 and IFM 63240, we prepared a fragmented DNA library by using a Nextera DNA sample preparation kit according to the manufacturer’s instructions (Illumina, San Diego, CA). The mean length of the libraries was 376–674 bp. We performed sequencing in a paired-end 2 × 150 bp mode on a MiSeq system (Illumina). For analyzing IFM 63666 and IFM 63768, we prepared a fragmented DNA library using KAPA HyperPlus Library Preparation Kit (Kapa Biosystems, Wilmington, MA) according to the manufacturer’s instructions. We performed sequencing in a paired-end 2 × 300 bp mode on a MiSeq system (Illumina).
Single-Nucleotide Variant Detection
To search for single-nucleotide polymorphisms (SNPs) between IFM 60814, the first isolate obtained, and IFM 63240–63243, the third set of isolates, we performed read-mapping and SNP detection using CLC Genomics Workbench (CLC bio, Aarhus, Denmark). In brief, we trimmed the reads from each isolate and mapped them to the Af293 reference genome. By comparing them with the Af293 sequence, we detected sites with SNPs in IFM 60814, IFM 63240, IFM 63241, IFM 63242, and IFM 63243. We also compared the sets of varied sites between IFM 60814 and IFM 63240–63243. We considered the unique sites common in IFM 63240–63243 to be the SNPs between the first isolate and the third set of isolates.
We cleaned the read datasets of IFM 63666 and IFM 63768 using Trimmomatic version 0.33 (17). We trimmed reads to generate de novo assembly of the draft genome of IFM 63666 using Platanus version 1.2. 4 with default parameters (18). We performed annotation of all predicted open reading frames of the draft genome using AUGUSTUS version 2.5.5 (19). For nucleotide differences between IFM 63666 and IFM 63768, we mapped the trimmed reads of IFM 63768 to the draft genome of IFM 63666 using SMALT version 0.7.6 (http://www.sanger.ac.uk/science/tools/smalt-0). We identified SNPs using SAMtools version 0.1.19–44428cd (20) and filtered with >10-fold coverage, >30 mapping quality, and 75% consensus using in-house scripts (21,22). We annotated the functional effect of SNPs with SnpEff version 4.1l (23).
Growth Test for Ergosterol-Related Inhibitors
We performed disk diffusion assay using the protocol described by Qiao et al. (24). We spread a 200-µL suspension (1 × 106 conidia/mL) of each of the tested isolates uniformly onto YGA (glucose 2%, yeast extract 0.5%, agar 2%, trace element). We impregnated blank paper disks 8 mm in diameter with 2.5 µg amphotericin B, 100 µg nystatin, or 40 µg lovastatin and placed the disks onto the center of the inoculated agar plates. We incubated the plates at 37°C and measured the diameters of the zones of inhibition 48 hours later. We performed each assay 3 times and calculated the mean diameter.
Ergosterol Quantification
We conducted total ergosterol extraction using the protocol described by Arthington-Skaggs et al. and Alcazar-Fuolietal (25,26). We cultured A. fumigatus isolates for 20 h in glucose minimal medium. We harvested mycelia by filtration, washed them with sterile water, and then dried and weighed them. We added 3 mL of 25% alcoholic potassium hydroxide solution to dried mycelia and mixed for 1 min, then incubated the mixture in an 80°C water bath for 1 h. After incubation, we added water and 3 mL of pentane and mixed for 3 min. We transferred the upper pentane layer to a clean glass tube for evaporation. We dissolved the dried samples in 1 mL of methanol and filtered it through a 0.22-µm pore size membrane filter. We analyzed ergosterol content using the Shimadzu LC-20A system (Shimadzu, Kyoto, Japan) with COSMOSIL 5C18-MS-II column (4.6 mm ID × 100 mm; Nacalai Tesque, Kyoto, Japan). We established a flow rate of 1 mL/min of acetonitrile to water (95:5 vol/vol). We used peak areas and heights recorded at a 254-nm wavelength in an RF-20Axs (Shimadzu) for quantification, expressing total ergosterol concentration as µg ergosterol per mg fungal dry weight. We repeated each experiment >3 times.
Construction of Mutant Erg6, Hmg1, and Cyp51A Expressed Strains
To construct transformants expressing erg6, hmg1, or cyp51A with the mutation, we cloned the alleles using the shuttle vector pPTR I (Takara Bio, Otsu, Japan) and GeneArt Seamless Cloning and Assembly Kit (Invitrogen, Tokyo, Japan). Primers used for the cloning are listed in Technical Appendix Table 2. We used genome DNAs of IFM 60814 (for wild-type [WT] gene) and IFM 63240 (for mutated allele) as templates to clone the alleles. The resultant plasmids were pPTRI-erg6WT, pPTRI-hmg1WT, pPTRI-cyp51AWT, pPTRI-erg6A350T, pPTRI-hmg1S269F, and pPTRI-cyp51AG448S, which we used for transformation of isolate IFM 60814. After selection with pyrithiamine, we verified the sequences of each gene of the candidate transformants by Sanger sequencing.
Multiazole Resistant Strains from a Single Patient
We recovered a total of 19 A. fumigatus isolates from 1 patient on 9 testing dates during September 2011–May 2016 (Tables 1, 2). Microsatellite analysis showed almost identical short tandem repeats across the isolates, derived from a genetically clonal background. The patient started treatment with VRCZ after the first isolation of A. fumigatus in September 2011. Whereas the first isolate (IFM 60814) was susceptible to antifungal drugs including ITCZ, VRCZ, and PSCZ, later isolates showed resistance to the azoles (MICs: ITCZ, 4 to >8; VRCZ, 8 to >8; PSCZ, 4 to >8). By sequencing the cyp51A gene, we found amino acid substitution G448S IFM 62916 (second isolation date), IFM63248 (fourth isolation date), IFM63595 (sixth isolation date), IFM63712 (seventh isolation date), IFM63713 (seventh isolation date), and IFM64139 (eighth isolation date). These isolates were most likely resistant to VRCZ as a result of the G448S mutation, which has been reported to confer VRCZ resistance in A. fumigatus. However, the multiazole resistance mechanism in the isolates with no G448S mutation remained unknown. The growth of isolate IFM 62916 was markedly impaired with glucose minimal medium and potato dextrose agar, whereas the isolates from the third isolation date also showed a moderately delayed growth compared with those of the first strain (Technical Appendix Figure 1).
Genome-Wide Sequence Comparison of Azole-Susceptible and Azole-Resistant Isolates
To gain insight into the multiazole resistance mechanism in the non-cyp51A strains, we sequenced the genomes of the isolates from the first (IFM 60814) and third (IFM 63240–63243) testing dates by next-generation sequencing. Compared with IFM 60814, IFM 63240–63243 commonly showed 7 nonsynonymous mutations, including 6 SNPs (4 amino acid substitutions and 2 nonsense mutations, which generate termination codon) and a 1-bp deletion resulting in a frame shift of the protein (Technical Appendix Table 2). The 6 genes with mutations included erg6, encoding sterol 24-C-methyltransferase and hmg1, encoding hydroxymethylglutaryl-CoA (HMG-CoA) reductase. The mutation of erg6 (A350T) resided in a Sterol_MT_C domain (PF08498) in the C-terminus, whereas the mutation of hmg1 (S269F) was located at the beginning of the sterol-sensing domain (PF12349) (Technical Appendix Figure 2). Because both of these genes are functionally related to the ergosterol biosynthesis pathway, we assumed that these mutations affect ergosterol biosynthesis and the consequential azole resistance in the strains.
We examined the rest of the isolates from the patient for mutations in the hmg1 and erg6 genes. Despite the presence of A350T, we identified E49K in the erg6 gene in several isolates, whereas Hmg1 S269F existed in all isolates tested (Tables 1, 2). These results indicate that all of the isolates showing multiazole resistance had mutations in both hmg1 and erg6 genes, regardless of the mutation in cyp51A gene.
Different Ergosterol Levels in Multiazole-Resistant Isolates
Figure 1
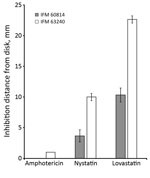
Figure 1. Growth inhibition by lovastatin and polyene drugs in azole-susceptible and azole-resistant Aspergillus fumigatus isolates from a patient in Japan. Growth inhibition tests were conducted by disk assay. IFM 60814 is a...
Figure 2
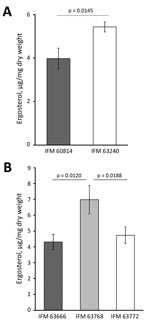
Figure 2. Total ergosterol content of azole-susceptible and azole-resistant Aspergillus fumigatus isolates in Japan. Ergosterol was quantified in the sets of IFM 60814, a susceptible isolate identified on the first date of testing,...
We investigated the phenotypes associated with the ergosterol biosynthesis pathway in IFM 63240, a representative of the isolates from the third testing day. Disk diffusion assay showed that this isolate was more sensitive to polyene drugs, amphotericin B, and nystatin, suggesting that ergosterol production might be differentially regulated (Figure 1). The ergosterol content in the cells, measured by HPLC, showed significant increase (p = 0.0145) in the third set of isolates compared with the first isolate (Figure 2, panel A). At the same time, we found no differences between the first and third isolates regarding sensitivity to terbinafine, which interferes with the early stage of ergosterol biosynthesis, and fenpropimorph, which inhibits ergosterol biosynthesis (data not shown). Of note, the isolate from the third set showed increased sensitivity to lovastatin, an inhibitor of HMG-CoA reductase (Figure 1).
Evaluation of Mutant erg6 and hmg1 by Transformation
Considering the different level of ergosterol biosynthesis in the third set of isolates, we wanted to determine whether the mutation in erg6 and/or hmg1 genes was responsible for the phenotypes (i.e., sensitivity to polyenes and resistance to azoles). To address this question, we constructed the isolates carrying these mutant alleles as well as the corresponding wild-type gene by introducing erg6A350T or hmg1S269F into the first strain (hereafter designated as 1st+erg6A350T and 1st+hmg1S269F). These mutant alleles were ectopically expressed in the transformed strains. We also constructed the strain carrying cyp51AG448S (1st+cyp51AG448S) for verification of the role of the mutation in azole resistance as previously reported (27). The MICs for azoles were determined in these strains (Table 4), showing that introducing erg6A350T or hmg1S269F did not affect susceptibility to azoles, whereas the cyp51AG448S conferred resistance to VRCZ.
Mutations in erg6 and hmg1 in Other Non-cyp51A Azole-Resistant Isolates
We studied whether the other non-cyp51A azole-resistant isolates possess mutations in erg6 or hmg1. Over the past decade, we have collected clinical azole-resistant A. fumigatus strains in Japan: 4 non-cyp51A azole-resistant isolates (IFM 62140, IFM 64258, IFM 64303, and IFM 63768) showing a VRCZ MIC of >8 (Table 5). Each isolate was from a patient who had a history of treatment with VRCZ before isolation. Although IFM 64303 has no mutations in erg6 and hmg1 genes, the other 3 isolates possessed different mutations in erg6 and/or hmg1. IFM 62140 had erg6A75S and hmg1F261_del, IFM 63768 had hmg1S269Y, and IFM 64258 had erg6V206A and hmg1F390Y, which were different from the pattern observed in IFM 63240. These results suggested a potential link between azole resistance and the mutations in erg6 and hmg1 genes. Notably, IFM 63768 and IFM 63240 had a mutation in Hmg1 at S269 in which serine was substituted by different amino acids (i.e., Hmg1S269Y and Hmg1S269F). To investigate the possibility that such mutations are just frequently occurring polymorphisms, we sequenced erg6 and hmg1 in 16 azole-susceptible isolates. We did not find mutation in erg6, although 1 of the 16 isolates, IFM 60901, had a hmg1H564Y mutation (Table 3). We did not find the mutations hmg1S269F/Y, hmg1F261_del, and hmg1F390Y in the azole-susceptible isolates. These results suggest that the mutations in erg6 and hmg1 found in the azole-resistant isolates are not mere polymorphisms in A. fumigatus but can be implicated in azole resistance.
Comparing Genome Sequences of Non-cyp51A Azole-Resistant Isolate and Corresponding Isolates
In our collection, an azole-susceptible isolate (IFM 63666) was isolated 2 months before isolation of azole-resistant IFM 63768 (hmg1S269Y) from the same patient. The 2 isolates shared the same short tandem repeats, indicating a clonal lineage. We then sequenced the genomes of IFM 63768 and IFM 63666. Comparing the genome sequences, we discovered only 1 nonsynonymous mutation other than hmg1S269Y in IFM 63768, which was I395M in Afu4g04290 encoding a putative histone H4 deacetylase protein. Although we could not see the effect of the mutation in Afu4g04290, this result supported the possibility that Hmg1S269Y is the determinant for azole resistance in this isolate. We compared ergosterol levels in the cells between IFM63768 and IFM 63666 and found an increased amount in IFM 63768 with hmg1S269Y (Figure 2, panel B). We isolated IFM 63772 from the same patient in addition to these isolates, and it showed moderate resistance to VRCZ, resistance to PSCZ, and no mutations in erg6 and hmg1 (Table 5). The amount of ergosterol in IFM 63772 was comparable to that of IFM 63666 (Figure 2, panel B).
The emergence of drug-resistant fungal strains has been on the rise in recent years, leading to a serious situation. Azole-resistant strains occurring during the course of therapy have been increasingly reported all over the world since the early discovery of an ITCZ-resistant strain (5). Some studies have shown that there are many azole-resistant A. fumigatus strains without mutation in cyp51A gene, and the molecular mechanisms underlying resistance in the strains remain unstudied (28–30). This finding indicates that many unclear molecular mechanisms related to azole resistance still exist.
In this study, we screened novel mutations conferring resistance by comparing the genomic sequences of susceptible and resistant strains isolated from the same patient. We found mutations in hmg1 and erg6, encoding enzymes Hmg1 and Erg6, respectively, which are involved in ergosterol biosynthesis. Hmg1 is a HMG-CoA reductase that catalyzes reduction of HMG-CoA to mevalonic acid and acts as a rate-limiting enzyme in ergosterol biosynthesis. Erg6 encodes sterol methyltransferase, which catalyzes the conversion of lanosterol to eburicol in A. fumigatus (26). The mutations of hmg1 we discovered were located in the sterol-sensing domain (SSD), responsible for the function of this enzyme. Thus, the mutation (hmg1S269F) might affect ergosterol biosynthesis efficiency. In fact, ergosterol levels in the strain bearing hmg1S269F or hmg1S269Y were increased, which led us to presume that such mutations altered the activity of Hmg1 and resulted in hyperaccumulation of ergosterol in the cells. Although further clarification is needed, one possibility is that larger amounts of azole drugs are required to inhibit growth of the cells with an increased level of ergosterol. Among the non-cyp51A azole-resistant strains we studied, all the strains bearing mutation in hmg1 showed resistance to multiple azoles. This finding also supported the idea that the ergosterol level is a determinant for the resistance against any class of azoles.
Because Hmg1 acts as a rate-limiting enzyme, we investigated the phenotypes associated with ergosterol biosynthesis in the hmg1 mutated strain. Polyene drugs such as amphotericin B and nystatin bind to ergosterol in the plasma membrane, leading to cell death by promoting membrane leakage. Of note, the strain with mutation in Hmg1 was more sensitive to these polyene drugs, compared with the strains without the mutation, likely because the azole-resistant strain has more ergosterol in the cell membrane, and polyene can increasingly access ergosterol and damage the membrane. We also found that the azole-resistant strain with Hmg1 mutation had increased susceptibility to lovastatin. Lovastatin is a competitive inhibitor of Hmg1, so the mutation may have increased the affinity to lovastatin. However, clarification for this will still be required in future studies.
Mutation of hmg1 has been reported in experiments designed to produce azole-resistant strains in the laboratory (31). In this study, the mutation in hmg1 was identified in clinical azole-resistant isolates. The mutation was discovered in several isolates with different genetic backgrounds, which strongly suggested the importance of the mutation in azole resistance.
To analyze functions of the amino acid substitutions in Cyp51A, Hmg1, and Erg6 identified here, mutant alleles (cyp51AG448S, hmg1S269F, and erg6A350T) were ectopically expressed in susceptible strains. Introducing mutated cyp51AG448S conferred azole resistance in the host strain even in the presence of wild-type cyp51A. On the other hand, mutations in hmg1 and erg6 did not confer azole resistance when the wild type was present. The amino acid substitution of Hmg1 is present in SSD, which is a conserved motif of membrane proteins involved in sterol sensing and acts on feedback regulation of the enzymatic reaction (32). Therefore, the mutated Hmg1 may be impaired in the feedback regulation. In the strain expressing hmg1S269F, it is considered that wild-type hmg1 can receive feedback inhibition, and thus the strain did not accumulate ergosterol and did not alter azole susceptibility. All mutations of hmg1 found in this study are present in SSD, suggesting that mutations in SSD are important for conferring azole resistance.
In this study, we discovered novel genetic changes related to azole resistance. Several non-cyp51A azole-resistant clinical isolates harbor the mutation in hmg1, which suggested that this possible resistance mechanism is prevalent in non-cyp51A strains. We proposed that multiazole drug resistance is imparted by a mutation in a protein other than the target protein of azole drugs. Future elucidation of the molecular mechanism of the hmg1 mutation will lead to a more complete understanding of the azole resistance mechanism in A. fumigatus.
Dr. Hagiwara is an associate professor with the Faculty of Life and Environmental Sciences, University of Tsukuba, Japan. His research interests include azole drug resistance mechanism and fungal secondary metabolite. Dr. Arai is research assistant professor with the Medical Mycology Research Center, Chiba University, Japan. His research interests include azole drug resistance mechanism, resistance gene detection method, and fungal secondary metabolite.
Acknowledgment
This research was supported by AMED under grant nos. JP18jm0110015 and JP18fk0108008.
References
- Shao PL, Huang LM, Hsueh PR. Recent advances and challenges in the treatment of invasive fungal infections. Int J Antimicrob Agents. 2007;30:487–95. DOIPubMedGoogle Scholar
- Meis JF, Chowdhary A, Rhodes JL, Fisher MC, Verweij PE. Clinical implications of globally emerging azole resistance in Aspergillus fumigatus. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150460. DOIPubMedGoogle Scholar
- Chowdhary A, Sharma C, Meis JF. Azole-resistant aspergillosis: epidemiology, molecular mechanisms, and treatment. J Infect Dis. 2017;216(suppl_3):S436–44. DOIPubMedGoogle Scholar
- Hagiwara D, Watanabe A, Kamei K, Goldman GH. Epidemiological and genomic landscape of azole resistance mechanisms in Aspergillus fungi. Front Microbiol. 2016;7:1382. DOIPubMedGoogle Scholar
- Warrilow AG, Parker JE, Price CL, Nes WD, Kelly SL, Kelly DE. In vitro biochemical study of cyp51-mediated azole resistance in Aspergillus fumigatus. Antimicrob Agents Chemother. 2015;59:7771–8. DOIPubMedGoogle Scholar
- Chowdhary A, Kathuria S, Xu J, Meis JF. Emergence of azole-resistant aspergillus fumigatus strains due to agricultural azole use creates an increasing threat to human health. PLoS Pathog. 2013;9:e1003633. DOIPubMedGoogle Scholar
- Fuhren J, Voskuil WS, Boel CH, Haas PJ, Hagen F, Meis JF, et al. High prevalence of azole resistance in Aspergillus fumigatus isolates from high-risk patients. J Antimicrob Chemother. 2015;70:2894–8. DOIPubMedGoogle Scholar
- Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, et al. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med. 2008;5:e219. DOIPubMedGoogle Scholar
- Vermeulen E, Lagrou K, Verweij PE. Azole resistance in Aspergillus fumigatus: a growing public health concern. Curr Opin Infect Dis. 2013;26:493–500. DOIPubMedGoogle Scholar
- Paul S, Diekema D, Moye-Rowley WS. Contributions of Aspergillus fumigatus ATP-binding cassette transporter proteins to drug resistance and virulence. Eukaryot Cell. 2013;12:1619–28. DOIPubMedGoogle Scholar
- Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, et al. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother. 2013;68:1486–96. DOIPubMedGoogle Scholar
- Hagiwara D, Miura D, Shimizu K, Paul S, Ohba A, Gonoi T, et al. A novel zn2-cys6 transcription factor atrr plays a key role in an azole resistance mechanism of Aspergillus fumigatus by co-regulating cyp51A and cdr1B expressions. PLoS Pathog. 2017;13:e1006096. DOIPubMedGoogle Scholar
- Camps SM, Dutilh BE, Arendrup MC, Rijs AJ, Snelders E, Huynen MA, et al. Discovery of a HapE mutation that causes azole resistance in Aspergillus fumigatus through whole genome sequencing and sexual crossing. PLoS One. 2012;7:e50034. DOIPubMedGoogle Scholar
- Gsaller F, Hortschansky P, Furukawa T, Carr PD, Rash B, Capilla J, et al. Sterol biosynthesis and azole tolerance is governed by the opposing actions of SrbA and the CCAAT binding complex. PLoS Pathog. 2016;12:e1005775. DOIPubMedGoogle Scholar
- Hagiwara D, Takahashi H, Watanabe A, Takahashi-Nakaguchi A, Kawamoto S, Kamei K, et al. Whole-genome comparison of Aspergillus fumigatus strains serially isolated from patients with aspergillosis. J Clin Microbiol. 2014;52:4202–9. DOIPubMedGoogle Scholar
- Hagiwara D, Watanabe A, Kamei K. Sensitisation of an azole-resistant Aspergillus fumigatus strain containing the Cyp51A-related mutation by deleting the SrbA gene. Sci Rep. 2016;6:38833. DOIPubMedGoogle Scholar
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. DOIPubMedGoogle Scholar
- Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24:1384–95. DOIPubMedGoogle Scholar
- Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33(Web Server):W465–W467.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. DOIPubMedGoogle Scholar
- Suzuki S, Horinouchi T, Furusawa C. Prediction of antibiotic resistance by gene expression profiles. Nat Commun. 2014;5:5792. DOIPubMedGoogle Scholar
- Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, et al. The molecular diversity of adaptive convergence. Science. 2012;335:457–61. DOIPubMedGoogle Scholar
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92. DOIPubMedGoogle Scholar
- Qiao J, Kontoyiannis DP, Wan Z, Li R, Liu W. Antifungal activity of statins against Aspergillus species. Med Mycol. 2007;45:589–93. DOIPubMedGoogle Scholar
- Arthington-Skaggs BA, Warnock DW, Morrison CJ. Quantitation of Candida albicans ergosterol content improves the correlation between in vitro antifungal susceptibility test results and in vivo outcome after fluconazole treatment in a murine model of invasive candidiasis. Antimicrob Agents Chemother. 2000;44:2081–5. DOIPubMedGoogle Scholar
- Alcazar-Fuoli L, Mellado E, Garcia-Effron G, Lopez JF, Grimalt JO, Cuenca-Estrella JM, et al. Ergosterol biosynthesis pathway in Aspergillus fumigatus. Steroids. 2008;73:339–47. DOIPubMedGoogle Scholar
- Krishnan Natesan S, Wu W, Cutright JL, Chandrasekar PH. In vitro-in vivo correlation of voriconazole resistance due to G448S mutation (cyp51A gene) in Aspergillus fumigatus. Diagn Microbiol Infect Dis. 2012;74:272–7. DOIPubMedGoogle Scholar
- Chowdhary A, Sharma C, Hagen F, Meis JF. Exploring azole antifungal drug resistance in Aspergillus fumigatus with special reference to resistance mechanisms. Future Microbiol. 2014;9:697–711. DOIPubMedGoogle Scholar
- Moye-Rowley WS. Multiple mechanisms contribute to the development of clinically significant azole resistance in Aspergillus fumigatus. Front Microbiol. 2015;6:70. DOIPubMedGoogle Scholar
- Wei X, Chen P, Gao R, Li Y, Zhang A, Liu F, et al. Screening and characterization of a non-cyp51A mutation in an Aspergillus fumigatus cox10 strain conferring azole resistance. Antimicrob Agents Chemother. 2016;61:e02101–16.PubMedGoogle Scholar
- Zhang J, van den Heuvel J, Debets AJM, Verweij PE, Melchers WJG, Zwaan BJ, et al. Evolution of cross-resistance to medical triazoles in Aspergillus fumigatus through selection pressure of environmental fungicides. Proc Biol Sci. 2017;284:20170635. DOIPubMedGoogle Scholar
- Theesfeld CL, Pourmand D, Davis T, Garza RM, Hampton RY. The sterol-sensing domain (SSD) directly mediates signal-regulated endoplasmic reticulum-associated degradation (ERAD) of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase isozyme Hmg2. J Biol Chem. 2011;286:26298–307. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: September 04, 2018
1These authors contributed equally to this article.
Table of Contents – Volume 24, Number 10—October 2018
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Akira Watanabe, Chiba University, Medical Mycology Research Center, 1-8-1 Inohana, Chuo-ku, Chiba City, Chiba, Japan
Top