Volume 32, Number 5—May 2026
Research
Zoonotic and Anthroponotic Plasmodium spp. Circulation between Wild Primates and Indigenous Community, Peruvian Amazon, 2007–2020
Cite This Article
Citation for Media
Abstract
Malaria transmission at the human–wildlife interface remains poorly characterized in the Amazon. We conducted a molecular survey of Plasmodium spp. in an Indigenous community (n = 141) and sympatric nonhuman primates (NHPs) (n = 341; 10 species) in the Peruvian Amazon during 2007–2020. By using nested or quantitative PCR (targeting cytb, cox3, and 18S rRNA genes) and sequencing, we estimated prevalence, parasite load, and genetic similarity. We detected Plasmodium in 43.3% of humans and 51.9% of NHPs. P. vivax/simium predominated in humans (42.1%), whereas P. brasilianum/malariae predominated in NHPs (24.6%). P. falciparum was rare in both hosts. Children <8 years of age showed higher parasite load than older persons. Bayesian phylogenies revealed >99.9% identity among human and NHP lineages, supporting shared Plasmodium lineages. NHP lineages showed low interannual variation. One third of human infections were asymptomatic. Our findings reveal hidden reservoirs and support integrating wildlife surveillance into Amazon malaria elimination strategies.
Malaria parasites of nonhuman primates (NHPs) are increasingly recognized for their zoonotic potential and complicate malaria control efforts (1). The first report of a zoonotic malaria parasite capable of infecting humans was Plasmodium knowlesi, which emerged as a major cause of human malaria in Southeast Asia (2). In South America, P. brasilianum (morphologically, genetically, and immunologically indistinguishable from P. malariae) (3) and P. simium (closely related to P. vivax) (4) parasites have been detected in NHPs and implicated in zoonotic infections (5,6).
The Amazon basin harbors high biodiversity and dense human–wildlife overlap, particularly within Indigenous territories (7). Indigenous Peoples and local communities live in malaria-endemic areas with limited access to health systems and maintain close contact with wildlife (8), which creates favorable conditions for cross-species transmission of pathogens, including malaria parasites (9). This convergence of factors amplifies the risk for zoonotic and anthroponotic malaria transmission. Reports of natural infections with P. vivax, P. simium, P. brasilianum, P. malariae, and P. falciparum parasites in all neotropical NHP families underscore their potential reservoir role (8,10–12). Furthermore, experimental infections of neotropical NHPs with zoonotic malaria parasites from Asia raise concerns about the vulnerability of NHPs to emerging zoonoses (13).
Despite global declines in malaria incidence (14), progress in the Amazon is hindered by ecologic complexity, vector diversity, and high rates of subpatent infections (15). A recent study conducted in a remote community in the northeastern Peruvian Amazon reported a P. vivax prevalence of 56.0% based on quantitative PCR (qPCR) results; nearly half of the infections were submicroscopic, highlighting the persistence of hidden reservoirs despite ongoing control interventions (16). Subclinical and mixed infections, common in malaria-endemic areas, evade standard diagnostics and are rarely captured in surveillance systems (17). Despite this fact, malaria-control efforts remain focused on P. falciparum and P. vivax parasites, and little attention is given to NHP-associated species such as P. brasilianum or P. malariae (18).
Detecting hidden transmission dynamics requires highly sensitive, specific molecular tools and ethical field strategies (19,20). In remote forest settings, community-based methods, such as analyzing blood from legal subsistence hunting, can provide access to hard-to-reach wildlife populations while respecting Indigenous sovereignty (21). We investigated the molecular epidemiology of Plasmodium infections in sympatric humans and NHPs within a remote Indigenous territory in the Peruvian Amazon. By using blood samples from 141 Indigenous inhabitants and 341 neotropical NHPs (from 10 species) hunted for subsistence during an 11-year long-range wildlife collection program, we applied multigene molecular diagnostics and phylogenetic analysis to assess parasite prevalence, diversity, and evidence of cross-species transmission.
Study Area
Figure 1
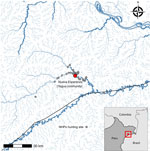
Figure 1. Geographic location of study of zoonotic and anthroponotic Plasmodiumspp. circulation between wild primates and Indigenous community, Peruvian Amazon, 2007–2020. Red dot indicates Yagua Indigenous community of Nueva Esperanza,...
We conducted this study in the Yavari-Mirin River basin, a remote upland forest region in the northeastern Peruvian Amazon. The area spans 107,000 hectares and includes a single Indigenous Yagua community, Nueva Esperanza, inhabited by 343 people in 2020. The community is located 302 km from the city of Iquitos and is surrounded by high biodiversity, including preserved populations of 14 NHP species (22). Malaria is endemic in the region; P. vivax is the most prevalent species. Villagers depend on subsistence activities (e.g., hunting, fishing, other forestry resources, and small-scale agriculture) and opportunistically trade wood, fish, wild meat, and agricultural products (Figure 1).
Ethical Considerations
All human study procedures were approved by the Ethics Committees of Universidad Peruana Cayetano Heredia (approval no. 102142), Universitat Autònoma of Barcelona (Comisión de Ética en la Experimentación Animal y Humana approval no. 4829), and Hospital Clínic of Barcelona (approval no. HCB/2019/1107) and authorized by local and regional authorities of Nueva Esperanza (approval no. 267–2019-GRL-DRSL/30.09.01). NHP sampling was approved by the Peruvian Forest and Wildlife Service (approval no. 258–2019-MINAGRI-SERFOR-DGGSPFFS) and the Institutional Animal Use Ethics Committee of the Universidad Peruana Cayetano Heredia. We exported wildlife samples under Servicio Nacional Forestal y de Fauna Silvestre permits (nos. 003258/SP, 003260/SP, 003568-SERFOR, and 003579-SERFOR).
Study Design and Blood Samples
In February 2020, we conducted a cross-sectional survey in Nueva Esperanza. We obtained whole blood samples through venipuncture from 141 participants (41.1% of the population) with informed consent, assent, or both. We referred participants with microscopic or clinical suspicion of malaria during the study to the community health center and treated them according to malaria management guidelines of Peru’s Ministry of Health (Supreme Decree no. 273–2025-MINSA).
In addition, we collected 17 samples from field workers; 5 were microscopically positive for P. vivax and had symptoms compatible with P. vivax infection; we molecularly analyzed samples of 3 of those workers and included results for sequence comparison only. Given the frequent malaria reports in Nueva Esperanza, field researchers received doxycycline as malaria chemoprophylaxis, in accordance with US Centers for Disease Control and Prevention (CDC) recommendations (23).
During 2007–2020, we collected dried blood spots (DBS) from 341 free-ranging NHPs (representing 10 species and 4 families) as part of a long-term wildlife monitoring program. Local hunters, trained in ethical sample collection, collected blood onto filter paper during postmortem handling of legally hunted animals. Species, sex, and date were recorded. The procedure was performed for all groups of mammals and birds (not only for wild NHPs) to avoid encouraging hunting of NHPs. The species sampled included 143 Poeppig’s woolly monkeys (Lagothrix lagothrica poeppigii), 57 large-headed capuchin monkeys (Sapajus macrocephalus), 43 black spider monkeys (Ateles chamek), 29 Ucayali bald uakaris (Cacajao calvus ucayalii), 21 red howler monkeys (Alouatta seniculus), 19 Humboldt’s white-fronted capuchin monkeys (Cebus albifrons), 16 monk saki (Pithecia monachus), 8 Ecuadorian squirrel monkeys (Saimiri macrodon), 4 coppery titi monkeys (Plecturocebus cupreus), and 1 brown-mantled tamarin (Leontocebus fuscicollis).
Microscopic Examination
We examined thick blood smears for 135 participants; 10 were positive and all confirmed by molecular assays. Because of low positivity and variable slide quality, we excluded microscopic examination results from analyses.
Molecular Diagnosis of Plasmodium Species
We extracted DNA from whole blood and DBS by using the AllPrep DNA/RNA Mini Kit (QIAGEN, https://www.qiagen.com). We collected all DBS from NHP in remote field conditions during 2007–2020, often with low DNA concentration and partial degradation. Therefore, we selected molecular targets to maximize sensitivity in cases of low parasitaemia and degraded material.
We detected parasites by using 2 nested PCRs and 2 commercial VIASURE qPCR diagnostic kits (Certest Biotec, https://www.certest.es) targeting mitochondrial cytochrome oxidase b (cytb), cytochrome c oxidase III (cox3), and 18S SSU rRNA genes. For quantitative analyses, we generated a standard curve by using serial dilutions of the kit’s Malaria Quantitative Standard (≈2 × 107 copies/µL) to estimate genome copy number. We converted cycle threshold values to DNA copies/µL and log10-transformed for statistical comparisons. Because of gene 18S rRNA copy number variability among Plasmodium species, our estimates reflect relative (not absolute) parasitemia.
Partial Amplification of cytb Gene
We performed nested PCR (nPCR) for cytb to detect Plasmodium spp. parasites in both humans and NHP samples, as described previously (24). PCRs included appropriate positive and negative controls and were processed under protocols that prevented contamination. We sequenced amplicons for species confirmation.
Validation of Plasmodium spp. Detection
Figure 2
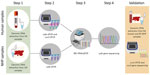
Figure 2. Workflow for molecular detection and validation of Plasmodium spp. infections in study of zoonotic and anthroponotic Plasmodiumspp. circulation between wild primates and Indigenous community, Peruvian Amazon,...
We validated a subset of 54 human and 81 NHPs samples at Leibniz Institute for Zoo and Wildlife Research (Berlin, Germany) (Figure 2) by using cox3 nPCR and sequencing. The cox3 nPCR uses genus-specific primers for the primary PCR, whereas the secondary PCR uses species-specific primers. The genus-specific primers were designed to avoid regions of high sequence similarity to human mitochondrial DNA, and each species-specific primer pair differs by >7 nucleotides at the 3′ end, which is critical for specificity. We used the oligonucleotides 5′-CTC GCC ATT TGA TAG CGG TTA ACC-3′ (forward) and 5′-CCT GTT ATC CCC GGC GAA CCT TC-3′ (reverse) as primers, as previously described (25), with some modifications: we performed first-round PCR in 25-μL reactions with 2 μM of each primer, 12.5 μL 2X MyTaq Mix (Meridian Bioscience, https://www.meridianbioscience.com), and 5 μL of template DNA. We conducted thermocycling: 96°C for 1 minute, followed by 40 cycles of 96°C for 10 seconds, 63°C for 1 minute, 72°C for 1 minute, and a final extension at 72°C for 10 minutes.
Nested reactions used 2.5 μL of 1:50-diluted PCR product, 4 μM primers, and 10 μL of 2X MyTaq Mix. We conducted cycling on P. falciparum and P. vivax/P. simium samples: 96°C for 1 minute; 30 cycles of 96°C for 10 seconds, 54°C for 1.5 minute, 72°C for 1 minute; and final extension at 72°C for 10 minutes. We applied the same protocol for P. brasilianum/P. malariae with 58 °C annealing. We gel-purified and sequenced amplicons for species confirmation. We performed nPCR separately for P. falciparum, P. vivax/P. simium, and P. brasilianum/P. malariae to generate sequences (Appendix Table 1).
Statistical Analysis
We analyzed data in R version 4.4.2 (The R Project for Statistical Computing, https://www.r-project.org). We estimated associations between symptoms and Plasmodium infection by using prevalence ratios and 95% CIs. We inferred parasite load from cycle threshold values converted to genome copy number (DNA copies/μL) by using a standard curve based on Plasmodium 18S rRNA qPCR and expressed these values as log10(DNA/μL + 1).
We assessed differences by age (<8 years vs. >8 years) and symptom status using Welch’s t-tests or Wilcoxon tests. We used linear regression models (Gaussian family, identity link) to evaluate age–symptom interactions.
We analyzed interannual NHPs prevalence by using a generalized linear model (binomial family, logit link) with host species and year as predictors. We assessed temporal stability by using coefficients of variation (CV), which we calculated as the SD divided by the mean prevalence per host–parasite pair; lower CV values indicated more stable infection patterns.
We compared patristic distances by using Kruskal-Wallis test and used pairwise Wilcoxon tests with Holm correction for multiple comparisons. We evaluated diagnostic agreement by using positive percent agreement (PPA), negative percent agreement (NPA), Cohen kappa coefficient, and percent concordance, using cytb nPCR sequencing as the reference standard. We defined statistical significance at α = 0.05.
Phylogenetic Analysis
We manually inspected chromatograms and corrected them for base-calling errors. We assembled consensus sequences in Geneious Prime version 2025.0.3 (Geneious, https://www.geneious.com) (26). We performed multiple sequence alignment in Geneious using the Clustal-Omega algorithm and visually trimmed ambiguous regions, resulting in final alignments of ≈776 bp of the mitochondrial cytb gene.
We inferred preliminary phylogenies by using the neighbor-joining method with 1,000 bootstrap replicates. We selected the best-fit nucleotide substitution model according to the Akaike information criterion in MEGA version 11.0.13 (27). We reconstructed final trees by using maximum-likelihood in FastTree version 2.1.11 (28) and Bayesian inference in MrBayes version 3.2.6 (29), applying the selected substitution model in both. Bayesian inference analyses consisted of 2 independent runs of 3 million generations, sampling every 1,000 generations, and a 25% burn-in.
We included reference Plasmodium sequences from GenBank and PlasmoDB to contextualize the evolutionary placement of lineages detected in humans and NHPs. Those sequences were P. falciparum, P. vivax (Sal-I, P01, PAM), P. simium, P. malariae, P. brasilianum, and rodent (P. chabaudi, P. vinckei, P. berghei, and P. yoelii) and avian (P. gallinaceum and P. relictum) Plasmodium species. The Apicomplexa parasite Toxoplasma gondii served as the outgroup. In addition, we incorporated 3 sequences from symptomatic field researchers, confirmed as P. vivax infections by microscopic examination and qPCR to assess their phylogenetic relationship with community-derived human and NHP infections.
Data Availability
Anonymized metadata will be available upon reasonable request. Sequence data are in GenBank (accession nos. PV769906–68 and PV786447–592) (Appendix Table 2). Protocols are available at https://www.protocols.io (Appendix).
Prevalence of Plasmodium spp. in Humans and NHPs
Among 141 human participants, we detected Plasmodium parasites in 35.5% (50/141) by qPCR and in 43.3% (61/141) by cytb nPCR. In 341 NHPs, prevalence reached 31.2% (69/221) by qPCR) and 51.9% (177/341) by cytb nPCR. Species-specific qPCR revealed P. vivax/P. simium in 30.5% (43/141) of samples, including 2 P. falciparum co-infections (1.4% [2/141]). Five samples were positive only at genus-level by qPCR. cytb nPCR sequencing confirmed P. vivax/P. simium in 42.1% (58/141) of participants, with single cases of P. falciparum and P. brasilianum/P. malariae (0.7% [1/141 each]). Species-level identification was successful in 98.4% (60/61) of nPCR-positive samples.
Among 110 persons for whom data were complete, 38 (34.6%) reported malaria-like symptoms (e.g., fever, headache, vomiting, nausea, pallor, or diarrhea). Of qPCR-positive patients, 16 (37.2%) were asymptomatic. Symptomatic persons were 3.8 times more likely to test positive (prevalence ratio 3.82 [95% CI 2.13–6.88]) (Table 1).
In NHPs, qPCR detected P. brasilianum/P. malariae in 25.8% (57/221), P. vivax/P. simium in 3.2% (7/221), and P. falciparum in 0.5% (1/221). cytb nPCR sequencing confirmed P. brasilianum/P. malariae in 24.6% (84/341), P. vivax/P. simium in 17.9% (61/341), and P. falciparum in 0.3% (1/341) (Table 2). Sequencing success was 82.5% (146/177).
Figure 3

Figure 3. Annual prevalence of Plasmodium spp. infections in wild nonhuman primate species detected in study of zoonotic and anthroponotic Plasmodiumspp. circulation between wild primates and Indigenous community,...
We observed no statistically significant interannual trend for P. brasilianum/P. malariae in NHPs (p = 0.414). However, host-specific variation emerged. Ucayali bald uakaris showed low interannual variation (CV 0.20, n = 29), suggesting stable P. brasilianum/P. malariae transmission. Poeppig’s woolly monkeys also showed sustained P. brasilianum/P. malariae prevalence (CV 0.26, n = 143). For P. vivax/P. simium, Humboldt’s white-fronted capuchin monkeys had the most consistent pattern (CV 0.36, n = 19), whereas black spider monkeys and large-headed capuchin monkeys showed more fluctuation (Figure 3; Appendix Table 3).
Parasite Load
Figure 4

Figure 4. Age-specific parasite load in humans based on quantitative PCR targeting for Plasmodium spp. 18S rRNA genes in study of zoonotic and anthroponotic Plasmodiumspp. circulation...
Participants (n = 141) included 84 women (59.6%) and 57 men (40.4%). Median age was 21 years (interquartile range 11–35, range 3–79 years); 9 participants (6.4%) were <8 years of age. Children <8 years of age had significantly higher relative parasite DNA levels inferred from qPCR (mean +SD log10 2.4 +1.1 vs. 0.9 +0.8; p = 0.005) than older persons (Figure 4). We found no significant differences in parasite load between symptomatic and asymptomatic participants. Linear modeling showed higher relative parasite loads in young children (β = 1.28, p = 0.020), independent of symptoms. We found no interaction between age and symptom status (p = 0.750).
Figure 5
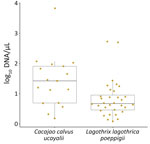
Figure 5. Comparison of parasite loads in 2 nonhuman primate species with Plasmodium brasilianum or P. malariae infections in study of zoonotic and anthroponotic Plasmodiumspp. circulation...
In NHPs, P. brasilianum/P. malariae infections had higher relative parasite DNA levels than P. vivax/P. simium (1.2 + 0.8 vs. 0.6 + 0.3; p<0.001). P. vivax in humans showed higher relative loads than P. vivax/P. simium in NHPs (p = 0.044), whereas P. vivax in humans and P. brasilianum/P. malariae in NHPs had similar densities (p = 0.851) (Table 3; Appendix Figure 2). Among NHPs, Ucayali bald uakaris exhibited significantly higher P. brasilianum/P. malariae relative loads than did Poeppig’s woolly monkeys (1.4 +0.9 vs. 0.8 +0.6; p = 0.020) (Figure 5).
Sequence and Phylogenetic Analysis
We analyzed a total of 209 partial cytb sequences, consisting of 60 human sequences, 3 sequences from fieldworkers, and 146 sequences from NHPs. Intraspecific and interspecific patristic distances based on cytb sequences revealed marked differences in genetic divergence among Plasmodium species across host groups (χ2 = 11.18 by Kruskal-Wallis test; degrees of freedom = 4, p<0.001). Human sequences had lower genetic divergence (mean 0.007) than did NHPs (mean 0.03; p<0.001). P. vivax/P. simium sequences from both hosts had very low divergence (mean 0.0007); P. brasilianum/P. malariae sequences showed even lower divergence (mean 0.0005, p<0.001). We found P. falciparum in 1 human and 1 NHP, with 99.6% identity.
Figure 6

Figure 6. Bayesian phylogeny of Plasmodium spp. from humans and nonhuman primates in study of zoonotic and anthroponotic Plasmodiumspp. circulation between wild primates and Indigenous community, Peruvian Amazon,...
Bayesian phylogenetics revealed 3 clades: P. vivax/P. simium, P. brasilianum/P. malariae, and P. falciparum. Most (96.7% [58/60]) human sequences and 41.8% (61/146) of NHP sequences clustered within the P. vivax/P. simium clade (Figure 6). All fieldworkers’ sequences also clustered within P. vivax/P. simium. Sequences clustering within the P. brasilianum/P. malariaeclade comprised 57.5% (84/146) of NHP and 1.7% (1/60) of human sequences. P. falciparum appeared in 1 human and 1 NHP. FastTree and neighbor-joining trees matched the Bayesian results, showing consistent clustering and no host-specific structuring (Appendix Figures 3, 4).
Validation of Plasmodium Detection
We reanalyzed a subset of 54 human and 81 NHP samples at Leibniz Institute for Zoo and Wildlife Research by using cox3 nPCR. In humans, we confirmed P. vivax/P. simium in 31.5% (17/54) and mixed infections in 11.1% (6/54). Laboratory concordance was 83.3% (kappa 0.66). In NHPs, we confirmed P. brasilianum/P. malariae in 19.8% (16/81) and P. vivax/P. simium in 16.1% (13/81). Agreement metrics were as follows: PPA 25.4%, NPA 95.2%, kappa 0.08. For P. brasilianum/P. malariae only, agreement metrics were PPA 44.0%, NPA 100.0%, and kappa 0.40. We consistently recovered major parasite lineages across laboratories and markers, indicating the robustness of detecting dominant parasite lineages despite low interlaboratory concordance for wildlife samples attributable to low DNA concentration and partial degradation (Appendix Figure 5).
The challenge facing infectious disease studies in free-ranging wild animals lies in the accessibility of biological material. Therefore, we implemented a culturally sensitive and sustainable surveillance approach on the basis of blood samples from discarded byproducts of legal subsistence hunting by Indigenous Peoples, respecting national laws and local practices and enabling long-term access to remote, well-preserved ecosystems, regions that are typically underrepresented in public health and zoonotic research (20).
Our results revealed a high prevalence of Plasmodium spp. in both humans and NHPs. P. vivax and P. simium were dominant in humans, whereas P. brasilianum and P. malariae predominated in NHPs and included sporadic P. falciparum co-infections. Parasite load decreased with age in humans, suggesting development of partial immunity from around 8 years of age, consistent with previous evidence of early adaptive immune response to P. vivax (30).
Nonetheless, one third of infections were asymptomatic, highlighting the role of subclinical carriage in sustaining silent transmission (31). In addition, similar findings in the remote community of Santa Emilia, in the northeastern Peruvian Amazon, indicated 44.0% submicroscopic P. vivax infections, underscoring the widespread presence of low-parasitemia reservoirs across the Peruvian Amazon (16). However, our study extends this evidence to sympatric wild primates, revealing a broader eco-epidemiologic interface of Plasmodium spp. circulation.
Assay performance varied by host and sample type, particularly in cases of low parasitaemia and long-term stored DBS from NHPs. Differences between qPCR (targeting the 18S rRNA gene) and nPCR (targeting the cytb gene) probably reflect gene copy number variation (32). Incorporating an independent cox3 nPCR improved confirmation of infection and detection of coinfections, enhancing resolution for subpatent infections in both hosts. However, large-scale implementation remains challenging because of structural barriers, limited access to healthcare, and reliance on suboptimal diagnostic tools, which contribute to underreporting of malaria cases and are critical obstacles to effective surveillance and control (33).
Molecular and phylogenetic analyses confirmed P. vivax/P. simium and P. brasilianum/P. malariae in both hosts. Although human sampling occurred in 2020 and NHP samples spanned 2007–2020, several human sequences matched NHP sequences collected more than a decade earlier, suggesting long-term maintenance of shared Plasmodium lineages across hosts, rather than recent spillover. Human P. vivax/P. simium sequences showed high mitochondrial similarity, consistent localized outbreaks, whereas greater diversity in NHPs indicates their role as long-term reservoirs.
Plasmodium spp. prevalence in NHPs ranged from 33% to 52% and exhibited species-specific variation. Ucayali bald uakaris (C. c. ucayalii), Poeppig’s woolly monkeys (L. l. poeppigii), Humboldt’s white-fronted capuchin monkeys (C. albifrons), black spider monkeys (A. chamek), and large-headed capuchin monkeys (S. macrocephalus) showed higher relative parasite DNA levels and stable infections, potentially reflecting differences in host susceptibility, immune tolerance, ecologic exposure, and evolutionary history (34). Low-grade chronic infections in NHPs might parallel asymptomatic infections in humans (35). Of note, the uakari (C. c. ucayalii) of Peru is classified as a vulnerable species because of its restricted distribution (36). This species has distinctive red facial vasculature and shows consistently higher relative parasitaemia than animals from sympatric taxa (37). Although the biologic basis of this pattern remains unclear, it may reflect species-specific ecologic or physiologic traits rather than host–parasite coevolution.
The single P. falciparum detection in a wild NHP (monk saki [P. monachus]), confirmed by 2 independent molecular assays, probably represents anthroponotic spillover. This finding underscores the permeability between human and animal malaria cycles and the need to monitor bidirectional transmission (10,11).
Our study provides longitudinal molecular evidence of Plasmodium spp. circulation in wild Amazonian NHPs (from 10 species) and 341 humans over an 11-year period. This approach, which integrated wildlife and human screening in parallel, provided a unique ecologic and temporal view of malaria dynamics in a remote Indigenous territory. Endemic P. brasilianum/P. malariae maintenance in wildlife contrasts with sporadic, low-parasitaemia P. vivax/P. simium detection. This pattern might reflect a complex eco-immunologic balance (34,35) in which P. vivax/P. simium circulates at undetectable levels in adapted NHP hosts, whereas P. brasilianum/P. malariae persists more overtly. Relative parasite DNA levels inferred from qPCR in P. vivax/P. simium–infected NHPs were ≈4 times lower than in local humans with low parasitaemia, reinforcing the need for sensitive and specific surveillance tools to detect cryptic reservoirs. However, that result should be interpreted cautiously, given that molecular estimates do not replace measurements acquired through microscopic examination.
Our findings carry important implications for malaria elimination. In Amazonian contexts, where humans and wildlife are closely linked through ecology and culture, traditional surveillance might miss important sources of transmission. Asymptomatic human infections and cryptic wildlife reservoirs add complexity to malaria ecology, reinforcing the need for One Health approaches that integrate wildlife monitoring and sensitive diagnostics.
One limitation of our study is that NHP samples were obtained opportunistically, taking advantage of waste materials from subsistence hunting; therefore, species representation reflects ecologic, behavioral, and seasonal factors, as well as hunter preferences influencing hunting practices, rather than the full primate community (38). Prevalence estimates thus should be interpreted carefully. We inferred parasite load from molecular data, and relative parasite DNA levels derived from qPCR should be interpreted cautiously because they do not replace parasitaemia measurements acquired through microscopic examination. We selected mitochondrial markers to optimize amplification from long-term storage DBS, low-parasitaemia samples, prioritizing sensitivity over fine-scale taxonomic resolution, but their limited resolution constrains discrimination between closely related parasites such as P. vivax and P. simium.
In conclusion, this study demonstrates that Plasmodium spp. transmission in the Peruvian Amazon involves overlapping human and NHPs reservoirs and that distinct parasite species predominate in each host. We show high prevalence of P. vivax/P. simium in humans and sustained circulation of P. brasilianum/P. malariae across multiple NHP species over more than a decade, confirming long-term maintenance of malaria parasites in wildlife. Detection of asymptomatic human infections and low-parasitaemia infections in NHPs highlights a hidden transmission layer that is largely missed by routine surveillance. Those findings indicate that malaria elimination efforts in Amazonian forest settings face additional challenges from cryptic reservoirs at the human–wildlife interface. Integrating sensitive molecular diagnostics and wildlife-informed surveillance within a One Health framework will be essential to reduce the risk for persistent transmission and reemergence in remote Indigenous territories.
Dr. Ulloa is a molecular eco-epidemiologist specializing in Plasmodium parasites at the human–wildlife interface. Her work integrates molecular diagnostics, population genomics, and evolutionary ecology to investigate zoonotic malaria transmission and host–parasite adaptation in the Amazon Basin.
Acknowledgments
We are very grateful to the people of Nueva Esperanza for their hospitality and their support in collecting biologic samples. We thank the Regional Directorate of Health and Management of Indigenous Affairs of the Regional Government of Loreto, the Iquitos Air Wing No. 5 of the Peruvian Air Force, the Municipality and the Health Medical Center of Islandia, the Comité Permanente de Atención Integral en Salud of the Scientific Society of San Fernando (Universidad Nacional Mayor de San Marcos), Campamento Universitario Multidisciplinario de Investigación y Servicio, volunteer students of human medicine and biology from the National University of the Peruvian Amazon, Winnie Michelle Contreras-Marmolejo, Arturo Mamani-Salce and the team of the Research Unit in Emerging Diseases and Climate Change of the Peruvian University Cayetano Heredia, Ricardo Gamboa, Katherina Alicia Viscaychipi, Milagritos Fernández Larrauri, and the team of physicians including Janet Cordori Carpio, Jackeline Magaly Tinoco Figueroa, Cristian Hipólito Andonaire Munaico, Augusto Gavino Escalante Candía, Saúl Javier Santiváñez Salazar, Sabina Mendivil Tuchia, and José Antonio Salinas Morales. We thank Karin Hönig for technical support. We thank Andree Valle-Campos for his advice on data analysis and graphics in R. We thank Alfredo Mayor and members of the Malaria Group of ISGlobal Barcelona of the Hospital Clinic of the University of Barcelona for their hospitality and support in carrying out the sample analysis.
This work was supported by ERANet-LAC (project no. ERANet17/HLH-0271), research projects (contract nos. 136-2018-FONDECYT, AC18/00054 Instituto de Salud Carlos, and Conselho Nacional de Desenvolvimento Científico e Tecnológico–400800/2019-5), and a training grant (no 2D43 TW007393) awarded to A.G.L. by the Fogarty International Center of the US National Institutes of Health. G.M.U. had 2 fellowships from Programa de Pós-graduação em Saúde e Produção Animal na Amazônia by Conselho Nacional de Desenvolvimento Científico e Tecnológico: a doctoral fellowship (GD modality; fellowship no. 140312/2020-0) and a doctoral sandwich fellowship (SWE modality; fellowship no. 201546/2020-5).
References
- Grigg MJ, Snounou G. Plasmodium simium: a Brazilian focus of anthropozoonotic vivax malaria? Lancet Glob Health. 2017;5:e961–2. DOIPubMedGoogle Scholar
- Barber BE, Rajahram GS, Grigg MJ, William T, Anstey NM. World malaria report: time to acknowledge Plasmodium knowlesi malaria. Malar J. 2017;16:135. DOIPubMedGoogle Scholar
- Bajic M, Ravishankar S, Sheth M, Rowe LA, Pacheco MA, Patel DS, et al. The first complete genome of the simian malaria parasite Plasmodium brasilianum. Sci Rep. 2022;12:19802. DOIPubMedGoogle Scholar
- Brasil P, Zalis MG, de Pina-Costa A, Siqueira AM, Júnior CB, Silva S, et al. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob Health. 2017;5:e1038–46. DOIPubMedGoogle Scholar
- Lalremruata A, Magris M, Vivas-Martínez S, Koehler M, Esen M, Kempaiah P, et al. Natural infection of Plasmodium brasilianum in humans: man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine. 2015;2:1186–92. DOIPubMedGoogle Scholar
- de Alvarenga DAM, Culleton R, de Pina-Costa A, Rodrigues DF, Bianco C Jr, Silva S, et al. An assay for the identification of Plasmodium simium infection for diagnosis of zoonotic malaria in the Brazilian Atlantic Forest. Sci Rep. 2018;8:86. DOIPubMedGoogle Scholar
- Pimm SL, Jenkins CN, Abell R, Brooks TM, Gittleman JL, Joppa LN, et al. The biodiversity of species and their rates of extinction, distribution, and protection. Science. 2014;344:
1246752 . DOIPubMedGoogle Scholar - de Abreu FVS, dos Santos E, Mello ARL, Gomes LR, de Alvarenga DAM, Gomes MQ, et al. Howler monkeys are the reservoir of malarial parasites causing zoonotic infections in the Atlantic forest of Rio de Janeiro. Fuehrer HP, editor. PLoS Negl Trop Dis. 2019;13:e0007906.
- Ferreira LM, Rezende HR, Fux B, De Alencar FEC, Loss AC, Buery JC, et al. Anopheles (Kerteszia) cruzii infected by Plasmodium in the Atlantic Forest indicates that the malaria transmission cycle is maintained even after howler monkeys’ population decline. Parasitol Res. 2022;121:3627–34. DOIPubMedGoogle Scholar
- Chaves A, Dolz G, Ibarra-Cerdeña CN, Núñez G, Ortiz-Malavasi EE, Bernal-Valle S, et al. Presence and potential distribution of malaria-infected New World primates of Costa Rica. Malar J. 2022;21:17. DOIPubMedGoogle Scholar
- Silva TRM, Barros FNL, Bahia M, Sampaio FD Junior, Santos SSF, Inoue LS, et al. Plasmodium vivax and Plasmodium falciparum infection in neotropical primates in the western Amazon, Brazil. Zoonoses Public Health. 2019;66:798–804. DOIPubMedGoogle Scholar
- Fuehrer HP, Campino S, Sutherland CJ. The primate malaria parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: genomic insights into distribution, dispersal and host transitions. Malar J. 2022;21:138. DOIPubMedGoogle Scholar
- Hang JW, Tukijan F, Lee EQH, Abdeen SR, Aniweh Y, Malleret B. Zoonotic malaria: non–Laverania Plasmodium biology and invasion mechanisms. Pathogens. 2021;10:889. DOIPubMedGoogle Scholar
- WHO. World malaria report 2022 [cited 2023 Mar 28]. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022
- Recht J, Siqueira AM, Monteiro WM, Herrera SM, Herrera S, Lacerda MVG. Malaria in Brazil, Colombia, Peru and Venezuela: current challenges in malaria control and elimination. Malar J. 2017;16:273. DOIPubMedGoogle Scholar
- Ramirez R, Torres K, Rodríguez P, Acosta C, Guzmán-Guzmán M, Llanos-Cuentas A, et al. High prevalence, genetic diversity and temporal differentiation of Plasmodium vivax in a remote hard-to-reach community from the Peruvian Amazon region. Am J Trop Med Hyg. 2025;113:990–6. DOIPubMedGoogle Scholar
- Almeida GG, Costa PAC, Araujo MDS, Gomes GR, Carvalho AF, Figueiredo MM, et al. Asymptomatic Plasmodium vivax malaria in the Brazilian Amazon: Submicroscopic parasitemic blood infects Nyssorhynchus darlingi. PLoS Negl Trop Dis. 2021;15:
e0009077 .PubMedGoogle Scholar - Cunha MG, Santos CS, Raiol M, Costa SPT, Ventura AMR, Póvoa MM, et al. Mixed Plasmodium malariae infections were underdetected in a malaria endemic area in the Amazon region, Brazil. Am J Trop Med Hyg. 2021;105:1184–6. DOIPubMedGoogle Scholar
- Estrada A, Garber PA, Rylands AB, Roos C, Fernandez-Duque E, Di Fiore A, et al. Impending extinction crisis of the world’s primates: why primates matter. Sci Adv. 2017;3:
e1600946 . DOIPubMedGoogle Scholar - Cáceres L, Calzada JE, Gabster A, Young J, Márquez R, Torres R, et al. Social representations of malaria in the Guna indigenous population of Comarca Guna de Madungandi, Panama. Malar J. 2017;16:256. DOIPubMedGoogle Scholar
- Jacob M, Medeiros Souza A, Martins de Carvalho A, Alves de Vasconcelos Neto CF, Tregidgo D, Hunter D, et al. Food biodiversity as an opportunity to address the challenge of improving human diets and food security. Ethnobiology and Conservation. 2023;12. DOIGoogle Scholar
- Mayor P, Pérez-Peña P, Bowler M, Puertas PE, Kirkland M, Bodmer R. Effects of selective logging on large mammal populations in a remote indigenous territory in the northern Peruvian Amazon. Ecol Soc. 2015;20:art36. DOIGoogle Scholar
- Centers for Disease Control and Prevention. Malaria. In: Brunette GW, Nemhauser JB, editors. CDC Yellow Book 2020: health information for international travel. New York: Oxford University Press; 2019.
- Ulloa GM, Greenwood AD, Cornejo OE, Monteiro FOB, Scofield A, Santolalla Robles ML, et al. Phylogenetic congruence of Plasmodium spp. and wild ungulate hosts in the Peruvian Amazon. Infect Genet Evol. 2024;118:
105554 . DOIPubMedGoogle Scholar - Isozumi R, Fukui M, Kaneko A, Chan CW, Kawamoto F, Kimura M. Improved detection of malaria cases in island settings of Vanuatu and Kenya by PCR that targets the Plasmodium mitochondrial cytochrome c oxidase III (cox3) gene. Parasitol Int. 2015;64:304–8. DOIPubMedGoogle Scholar
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. DOIPubMedGoogle Scholar
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. DOIPubMedGoogle Scholar
- Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50. DOIPubMedGoogle Scholar
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. DOIPubMedGoogle Scholar
- Ome-Kaius M, Kattenberg JH, Zaloumis S, Siba M, Kiniboro B, Jally S, et al. Differential impact of malaria control interventions on P. falciparum and P. vivax infections in young Papua New Guinean children. BMC Med. 2019;17:220. DOIPubMedGoogle Scholar
- Villasis E, Garcia Castillo SS, Guzman M, Torres J, Gomez J, Garro K, et al. Epidemiological characteristics of P. vivax asymptomatic infections in the Peruvian Amazon. Front Cell Infect Microbiol. 2022;12:
901423 . DOIPubMedGoogle Scholar - Echeverry DF, Deason NA, Davidson J, Makuru V, Xiao H, Niedbalski J, et al. Human malaria diagnosis using a single-step direct-PCR based on the Plasmodium cytochrome oxidase III gene. Malar J. 2016;15:128. DOIPubMedGoogle Scholar
- Ministerio de Salud del Perú. Análisis de situación de salud de los pueblos indígenas de la Amazonía, viviendo en el ámbito de las cuatro cuencas y el Río Chambira. 2020 Dec 1 [cited 2025 May 24]. https://www.gob.pe/institucion/minsa/informes-publicaciones/1893891-analisis-de-situacion-de-salud-de-los-pueblos-indigenas-de-la-amazonia-viviendo-en-el-ambito-de-las-cuatro-cuencas-y-el-rio-chambira
- Monteiro EF, Fernandez-Becerra C, Araujo MDS, Messias MR, Ozaki LS, Duarte AMRC, et al. Naturally acquired humoral immunity against malaria parasites in non-human primates from the Brazilian Amazon, Cerrado and Atlantic Forest. Pathogens. 2020;9:525. DOIPubMedGoogle Scholar
- de Assis GMP, de Alvarenga DAM, Costa Pereira MO, Sánchez-Arcila JC, de Pina Costa A, de Souza JC Junior, et al. Profiling humoral immune response against pre-erythrocytic and erythrocytic antigens of malaria parasites among neotropical primates in the Brazilian Atlantic Forest. Front Cell Infect Microbiol. 2021;11:
678996 . DOIPubMedGoogle Scholar - Ennes Silva F, Valsecchi do Amaral J, Roos C, Bowler M, Röhe F, Sampaio R, et al. Molecular phylogeny and systematics of bald uakaris, genus Cacajao (Primates: Pitheciidae), with the description of a new species. Mol Phylogenet Evol. 2022;173:
107509 . DOIPubMedGoogle Scholar - Mayor P, Mamani J, Montes D, González-Crespo C, Sebastián MA, Bowler M. Proximate causes of the red face of the bald uakari monkey (Cacajao calvus). R Soc Open Sci. 2015;2:
150145 . DOIPubMedGoogle Scholar - Rosin C, Swamy V. Variable density responses of primate communities to hunting pressure in a western Amazonian River Basin. Neotrop Primates. 2013;20:25–31. DOIGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: April 28, 2026
Table of Contents – Volume 32, Number 5—May 2026
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Gabriela M. Ulloa, Grupo de Enfermedades Infecciosas Re-emergentes, Universidad Científica del Sur, Crta. Panamericana sur km 19, Lima 15067, Peru
Top